







Rapport d'information n° 708 (2020-2021) de Mmes Annie DELMONT-KOROPOULIS et Véronique GUILLOTIN , fait au nom de la commission des affaires sociales, déposé le 23 juin 2021
Disponible au format PDF (1,4 Moctet)
Synthèse du rapport (562 Koctets)
-
LISTE DES PRINCIPALES PROPOSITIONS
-
AVANT-PROPOS
-
I. RENFORCER LA SOUVERAINETÉ SANITAIRE
FRANÇAISE ET EUROPÉENNE
-
A. RÉUNIR LES CONDITIONS D'UNE
RECONQUÊTE INDUSTRIELLE
-
B. DONNER À LA RECHERCHE LES MOYENS DE
VALORISER SES RÉSULTATS
-
A. RÉUNIR LES CONDITIONS D'UNE
RECONQUÊTE INDUSTRIELLE
-
II. DÉVELOPPER L'ACCÈS DES PATIENTS
À L'INNOVATION EN PRENANT RÉSOLUMENT LE VIRAGE DE LA
MÉDECINE PERSONNALISÉE
-
A. PERMETTRE À LA FRANCE DE RETROUVER SON
ATTRACTIVITÉ POUR ACCUEILLIR LA RECHERCHE CLINIQUE
-
1. Moderniser l'examen des demandes d'essais
cliniques
-
a) Un bilan en demi-teinte du raccourcissement des
délais d'autorisation
-
b) Des essais cliniques précoces
insuffisamment accueillis en France
-
c) Une expertise parfois critiquée
-
d) Des CPP embolisés par l'examen des
études observationnelles
-
e) Prendre le virage des essais adaptatifs et des
études post-commercialisation
-
a) Un bilan en demi-teinte du raccourcissement des
délais d'autorisation
-
2. Rénover le modèle de financement
de la recherche clinique
-
1. Moderniser l'examen des demandes d'essais
cliniques
-
B. REPLACER LE PATIENT AU CoeUR DE
L'INNOVATION
-
A. PERMETTRE À LA FRANCE DE RETROUVER SON
ATTRACTIVITÉ POUR ACCUEILLIR LA RECHERCHE CLINIQUE
-
I. RENFORCER LA SOUVERAINETÉ SANITAIRE
FRANÇAISE ET EUROPÉENNE
-
TRAVAUX DE LA COMMISSION
-
LISTE DES PERSONNES ENTENDUES
N° 708
SÉNAT
SESSION ORDINAIRE DE 2020-2021
Enregistré à la Présidence du Sénat le 23 juin 2021
RAPPORT D'INFORMATION
FAIT
au nom de la commission des affaires sociales (1) sur l' innovation en santé ,
Par Mmes Annie DELMONT-KOROPOULIS et Véronique GUILLOTIN,
Sénatrices
(1) Cette commission est composée de : Mme Catherine Deroche , présidente ; M. Jean-Marie Vanlerenberghe , rapporteur général ; M. Philippe Mouiller, Mme Chantal Deseyne, MM. Alain Milon, Bernard Jomier, Mme Monique Lubin, MM. Olivier Henno, Martin Lévrier, Mmes Laurence Cohen, Véronique Guillotin, M. Daniel Chasseing, Mme Raymonde Poncet Monge , vice-présidents ; Mmes Florence Lassarade, Frédérique Puissat, M. Jean Sol, Mmes Corinne Féret, Jocelyne Guidez , secrétaires ; Mme Cathy Apourceau-Poly, M. Stéphane Artano, Mme Christine Bonfanti-Dossat, MM. Bernard Bonne, Patrick Boré, Laurent Burgoa, Jean-Noël Cardoux, Mmes Catherine Conconne, Annie Delmont-Koropoulis, Élisabeth Doineau, MM. Alain Duffourg, Jean-Luc Fichet, Mmes Laurence Garnier, Frédérique Gerbaud, Pascale Gruny, M. Xavier Iacovelli, Mmes Corinne Imbert, Annick Jacquemet, Victoire Jasmin, Annie Le Houerou, M. Olivier Léonhardt, Mmes Viviane Malet, Colette Mélot, Michelle Meunier, Brigitte Micouleau, Annick Petrus, Émilienne Poumirol, Catherine Procaccia, Marie-Pierre Richer, Laurence Rossignol, M. René-Paul Savary, Mme Nadia Sollogoub, M. Dominique Théophile .
LISTE DES PRINCIPALES PROPOSITIONS
___________
Proposition n° 1 : développer en France une filière du médicament biologique sur l'ensemble de la chaîne de valeur, de la recherche à la production.
Proposition n° 2 : soutenir la localisation en France et en Europe de la production pharmaceutique par des mécanismes de prix garantissant des marges aux producteurs et par des dispositifs fiscaux incitatifs.
Proposition n° 3 : assouplir la progression de l'enveloppe « médicaments » au sein de l'Ondam afin de soutenir la production française.
Proposition n° 4 : consolider le rôle de la BPI dans le soutien à l'innovation en santé par un accroissement des investissements en capital-risque.
Proposition n° 5 : rendre accessibles sur les marchés français des montants d'investissements majeurs par une meilleure approche du risque et des fonds d'investissements drainant plus fortement les produits d'épargne.
Proposition n° 6 : amplifier la politique de sites en identifiant un ou deux sites prioritaires en France pour constituer des clusters d'envergure internationale soutenus par des financements publics massifs.
Proposition n° 7 : ouvrir la possibilité pour le mandataire unique de se transformer en un « propriétaire unique » disposant d'un mandat élargi et d'une autonomie renforcée pour la cession d'actifs et la conclusion de partenariats, sous réserve du respect d'une charte de principes partagés et d'un accord sur la répartition des revenus nets, validés par les copropriétaires dès la désignation du mandataire.
Proposition n° 8 : professionnaliser l'activité de valorisation conduite par les SATT dans le domaine de la santé en s'appuyant sur un benchmark des pratiques de transfert qui déboucherait sur un guide des bonnes pratiques pour l'élaboration des contrats de licence.
Proposition
n° 9
: remédier
au sous-financement structurel de la recherche biomédicale par :
- le doublement de la part des crédits de la mission
interministérielle « Recherche et enseignement
supérieur » dédiés à la recherche en
biologie-santé ;
- l'identification de quelques secteurs
à haut potentiel et stratégiques dans le domaine de la
santé en faveur desquels serait priorisé l'accès aux
financements publics.
Proposition n° 10
: créer
une agence de l'innovation en santé sous la forme d'un service à
compétence nationale placé sous l'autorité du ministre de
la santé, chargée des missions suivantes :
- définir, à partir d'un
horizon scanning
, une
stratégie de spécialisation sur les segments de recherche
porteurs de l'innovation en santé qui devront être soutenus
prioritairement pour répondre aux besoins du système de
santé ;
- attribuer une présomption d'innovation
à des développements et thérapies s'inscrivant dans ces
segments prioritaires afin de faciliter le lancement d'un essai clinique ou un
accès rapide au marché ;
- constituer un guichet
unique pour le dépôt centralisé d'un seul et même
dossier de candidature aux appels d'offres en faveur de l'innovation en
santé ;
- simplifier et harmoniser les procédures de
transfert de propriété intellectuelle par l'élaboration
d'un guide de bonnes pratiques.
Proposition n° 11
: moderniser
le fonctionnement des CPP par :
- le transfert de l'examen des
RIPH de catégorie 3 à un comité d'éthique
spécialisé sur les recherches non interventionnelles et la
protection des données de santé personnelles ;
- le
renforcement de la formation de leurs membres aux mécanismes des
innovations de rupture ;
- la constitution, au plus tard avant la
fin 2021, sous l'égide de la CNRIPH, d'un annuaire d'experts selon
différentes aires thérapeutiques à la disposition des CPP,
avec publication de leurs déclarations d'intérêts ;
- l'assouplissement, sous le contrôle de la CNRIPH qui serait
dotée d'un déontologue de l'expertise sanitaire, des conditions
de mobilisation de l'expertise pertinente en s'appuyant, à cet
égard, sur un
benchmark
des pratiques dans d'autres pays
européens en matière de prévention des conflits
d'intérêts ;
- la rénovation des modes
d'indemnisation des membres et experts des CPP pour valoriser leur engagement,
notamment en reconnaissant l'investissement des présidents et
vice-présidents ;
- la mise en place d'un audit
indépendant et périodique des CPP conditionnant le renouvellement
de leur agrément.
Proposition n° 12 : inscrire dans la loi un statut des comités d'éthique de la recherche chargés d'examiner les protocoles de recherche n'ayant pas de finalité biologique et médicale et clarifier les méthodologies de référence applicables à ces recherches.
Proposition n° 13 : prendre le virage des essais adaptatifs, des études post-AMM et de la collecte de données en vie réelle dans un objectif d'amélioration continue de la qualité des soins.
Proposition n° 14 : mettre en place une procédure accélérée ( fast-track ) pour permettre l'accès des chercheurs et des promoteurs et investigateurs d'essais cliniques aux données de santé du Health Data Hub pour évaluer l'ensemble des effets d'une thérapeutique.
Proposition n° 15 : étudier la faisabilité de la transposition du dispositif de passerelles soin/recherches de l'article 18 du projet de loi relatif à la bioéthique à la réutilisation de données de santé dans le cadre de programmes de recherche.
Proposition n° 16 : créer des registres nationaux de candidats à des essais cliniques sur de grands enjeux thérapeutiques.
Proposition n° 17 : augmenter l'enveloppe « recherches-inclusions » au sein des Merri à hauteur de 25 % de la dotation socle.
Proposition n° 18 : confier aux fédérations d'établissements de santé et aux organisations représentatives des promoteurs le soin d'établir des référentiels de coûts et de surcoûts et des méthodologies de calcul afin d'accélérer la négociation de l'annexe financière de la convention unique.
Proposition n° 19 : étendre l'éligibilité au CIR à l'ensemble des étapes de faisabilité (démarches pour trouver des patients, évaluation de la disponibilité des centres, monitoring ...) et de mise en place des essais cliniques (choix du pays où se dérouleront les essais, démarches règlementaires et d'éthique, formation des personnels des centres...).
Proposition n° 20 : améliorer le traitement des dossiers d'évaluation en vue de la prise en charge afin de garantir un accès rapide aux innovations.
Proposition n° 21 : débloquer les restrictions à la prise en charge de tests génétiques en oncologie.
Proposition n° 22 : permettre l'inscription conditionnelle sur la liste en sus de thérapies innovantes avec ASMR IV, correspondant aux innovations de rupture.
Proposition n° 23 : lever la nécessité de comprendre des patients français pour une étude soumise à la HAS en vue d'une prise en charge.
Proposition n° 24 : engager un plan d'action pour favoriser le développement en France d'organismes notificateurs.
Proposition n° 25 : privilégier l'hébergement des données de santé par un opérateur national ou européen.
Proposition n° 26 : assurer l'interopérabilité des données de santé collectées par les établissements de santé pour faciliter leur exploitation dans le Health Data Hub .
Proposition n° 27 : renforcer le suivi en vie réelle pour l'évaluation des médicaments faisant l'objet d'une prise en charge conditionnelle.
AVANT-PROPOS
Dans le cadre d'une mission d'information sur l'innovation en santé, la commission a dressé le bilan de la mise en oeuvre des mesures du 8 e conseil stratégique des industries de santé 1 ( * ) (CSIS), et préparé le prochain CSIS dont les orientations sont annoncées pour la fin du mois de juin 2021, en identifiant les principaux freins règlementaires, financiers, organisationnels mais aussi culturels qui demeurent pour stimuler l'innovation en santé en France et faciliter l'accès aux thérapies innovantes.
• Au travers d'auditions et de tables rondes qui ont rassemblé près de 60 personnes mais aussi de nombreuses contributions recueillies sur un temps resserré d'un mois et demi, les rapporteures ont consulté une grande variété d'acteurs du secteur, de la recherche académique et hospitalière à l'industrie, en passant par les acteurs institutionnels, des représentants de pôles de compétitivité en santé ou encore de start-ups en santé et de biotechs .
• Si environ 80 % des mesures du CSIS de juillet 2018 ont été mises en oeuvre , en particulier une refonte majeure du système d'accès précoce , certaines réformes clés restent à quai, dont celle de l' évaluation du médicament pour mieux l'adapter aux nouveaux mécanismes de l'innovation, et celle de l' expertise .
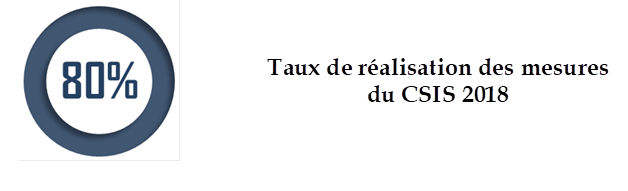
• La crise sanitaire liée à la covid-19 a, en outre, montré qu'une politique des petits pas n'était pas adaptée au rythme des innovations dans le secteur de la santé, qui a connu au cours de la dernière décennie une accélération spectaculaire , ni à l'ampleur des bouleversements induits par ces innovations sur le système de santé. La crise sanitaire a ainsi été particulièrement riche d'enseignements sur la situation de notre pays en matière d'innovation en santé, en servant à la fois :
- d' accélérateur d'évolutions favorables à cette innovation , au travers notamment de la multiplication de procédures accélérées - dites de « fast-track » - pour l'autorisation d'essais cliniques ou encore la mise sur le marché de vaccins ou de traitements, mais aussi de l'essor de la télémédecine , de la télésurveillance et d'autres innovations organisationnelles structurantes pour la continuité du parcours de soin entre l'hôpital et la ville ;
- de révélateur de faiblesses françaises préoccupantes . La blessure d' ego française dans la course aux vaccins contre la covid-19, avec le triomphe de deux biotechs américaine (Moderna) et allemande (BioNTech) qui tranche avec l'échec initial de Sanofi, n'est finalement qu'un des symptômes de ce qui s'apparente de plus en plus à un déclassement de notre pays dans le développement et la production de thérapies innovantes. La France ne s'est pas en effet emparée de l'innovation en santé comme facteur de croissance et de compétitivité, en sous-estimant très largement des tendances lourdes, dont l' effacement progressif des « frontières » entre recherche fondamentale et recherche clinique et appliquée avec l'essor de la recherche translationnelle 2 ( * ) , le besoin d'investissements massifs pour prendre le virage des biotechnologies et conserver dans le giron national des pépites innovantes, ou encore la délocalisation des capacités de production en principes actifs . En résulte une indépendance sanitaire française sérieusement entamée .
• Le 9 e CSIS s'inscrit, en outre, dans un moment clé pour refonder l'écosystème français et européen de l'innovation en santé , alors que des réformes d'ampleur prennent corps au niveau européen avec la création d'une agence européenne de réponse aux crises sanitaires, l'entrée en vigueur prochaine 3 ( * ) du règlement européen sur les essais cliniques de médicaments 4 ( * ) et l'entrée en vigueur depuis le 26 mai 2021 du règlement européen sur les dispositifs médicaux 5 ( * ) . À l'heure où les États-Unis et la Chine renforcent leur position de leaders dans le développement et la production de thérapies innovantes, ce cadre règlementaire européen accentue l' environnement concurrentiel dans lequel notre pays doit trouver sa place pour s'imposer comme moteur de l'innovation en santé dès ses phases les plus précoces, afin d'attirer les investissements stratégiques sur l'ensemble de la chaîne de valeur du médicament . Ce dernier CSIS de la mandature en cours doit donc être l'occasion d'un véritable saut qualitatif dans le développement de l'accès aux thérapies innovantes en France, en prenant résolument le virage de la médecine personnalisée .
Feuille de route du CSIS 2021
Le président de la République a fixé au CSIS 2021 un objectif ambitieux : faire de la France une nation leader en matière d'industrie et d'innovation en santé .
Pour établir un véritable et ambitieux schéma d'orientation « Santé-Innovation 2030 », cinq priorités ont été fixées :
- assurer une recherche fondamentale d'excellence et interdisciplinaire , capable d'alimenter l'innovation d'un flot continu, mais aussi assurer une continuité de la recherche fondamentale à la recherche clinique ;
- catalyser l'innovation : la France est riche de nombreuses jeunes entreprises innovantes ; le CSIS 2021 sera l'occasion de poser les jalons d'un dispositif permettant de sécuriser ces innovations en santé, d'améliorer l'accès au financement et faciliter le maintien du développement dans notre pays, y compris durant les phases risquées et à forte intensité capitalistique ;
- améliorer l'accès au marché des produits innovants afin de les rendre disponibles plus tôt pour les patients et renforcer l'intégration de ces innovations dans le parcours de soin ;
- soutenir l'industrialisation des produits dans la prolongation des objectifs de relocalisation des sites de production poursuivis dans le cadre de France Relance, pour disposer de capacités de production pharmaceutique suffisantes et permettre aux innovations d'être développées et produites en France ;
- développer et faire émerger les formations initiales et tout au long de la vie nécessaires à disposer des compétences pour réaliser les objectifs proposés.
Source : Extrait du communiqué de presse du 11 février 2021 de M. Olivier Véran et Mmes Frédérique Vidal et Agnès Pannier-Runacher.
I. RENFORCER LA SOUVERAINETÉ SANITAIRE FRANÇAISE ET EUROPÉENNE
A. RÉUNIR LES CONDITIONS D'UNE RECONQUÊTE INDUSTRIELLE
La France reste marquée par des priorités industrielles historiquement tournées vers l'énergie nucléaire, l'armement et l'aéronautique quand d'autres pays, comme les États-Unis et le Royaume-Uni, ont très tôt investi massivement dans l'innovation en santé. La crise sanitaire a remis en perspective le secteur de la santé comme un élément stratégique de souveraineté.
1. Prendre le virage des biotechnologies, tout en consolidant la filière du médicament mature
Alors que la France restait en position de leader européen en matière de production pharmaceutique de 1995 à 2008, la décennie qui s'est achevée a montré un déclassement de la France dans le secteur, passée au 4 e rang derrière la Suisse, l'Allemagne et l'Italie.
Production pharmaceutique en Europe : 10 principaux pays producteurs en 2017
en millions d'euros
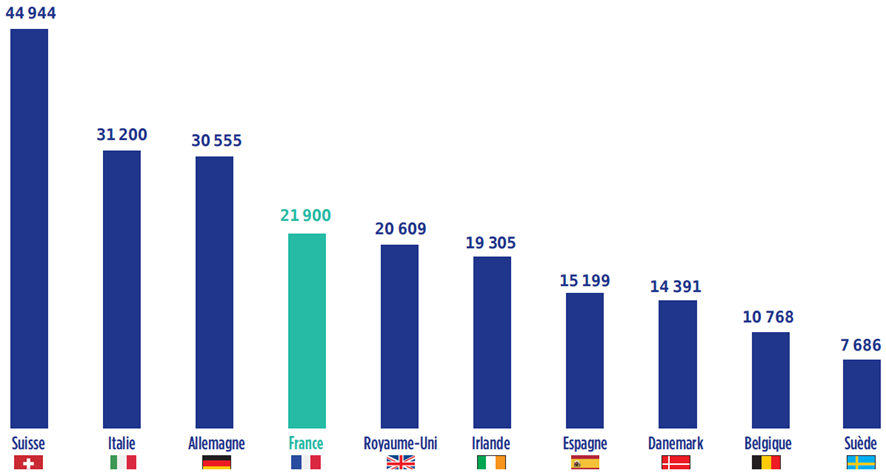
Source : EFPIA
a) Affirmer les biotechnologies comme priorité de développement de la production pharmaceutique
• Le dernier CSIS avait érigé en mesure phare la création d'un pôle d'excellence mondial en France dans le domaine des biotechnologies . Le bilan de cette mesure est jugé très mitigé par les acteurs auditionnés qui pointent au contraire un décrochage dans ce domaine. L'outil industriel français demeure aujourd'hui majoritairement consacré à la production de médicaments anciens.
Évolution de la production pharmarceutique,
chimique et biologique,
des principaux pays producteurs en Europe entre 2004
et 2014
en milliards d'euros
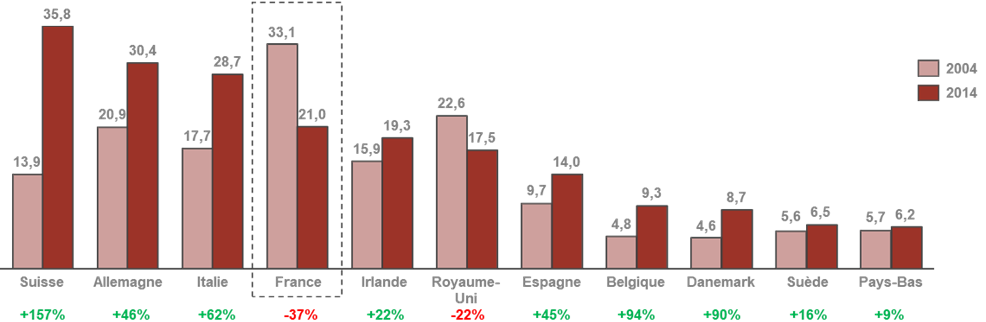
Source : AEC Partners, Cartographie de la bioproduction en France , rapport pour le Leem, janvier 2018
• Le secteur des bioproductions conserve aujourd'hui une place limitée dans la production pharmaceutique française : il ne représente que 32 sites sur 271 sites de production en France, et 8 500 emplois , pour une production par ailleurs consacrée à 60 % à la production de vaccins.
Ce positionnement résolu sur les biotechnologies nécessite une approche globale : il s'agit ici d'appréhender l'ensemble de la chaîne de valeur, de la recherche à la production.
Sur ce constat, les acteurs de la filière ont adopté en décembre 2020 un plan d'action visant à faire de la France le leader européen de la bioproduction à l'horizon 2030. Une structure de pilotage scientifique et industriel, « l'Alliance France bioproduction » a été ainsi constituée afin de structurer les actions de développement de la filière . La feuille de route comporte différents éléments de soutien à la recherche, de renforcement de la compétitivité mais aussi de consolidation d'un réseau d'intégrateurs à finalité industrielle. La filière s'est ainsi fixée un objectif : porter en dix ans la part de produits biologiques approuvés par l'Agence européenne des médicaments et fabriqués en France de 5 % à 20 %.
Proposition n° 1 : développer en France une filière du médicament biologique sur l'ensemble de la chaîne de valeur, de la recherche à la production.
Ces initiatives de la filière trouvent un soutien financier de la part des pouvoirs publics qui partagent le même objectif de faire de la France un leader : le Gouvernement, à travers le programme des investissements d'avenir (PIA) et le plan « France Relance » a ainsi lancé une stratégie d'accélération « biothérapies et bioproduction de thérapies innovantes ». Un « défi biomédicament » a également été lancé et doté de 30 millions d'euros financés par le fonds pour l'innovation.
Sites de bioproduction recensés et qualifiés* en France et majoritairement français
Seulement 3 appartenant à des groupes d'origine internationale**
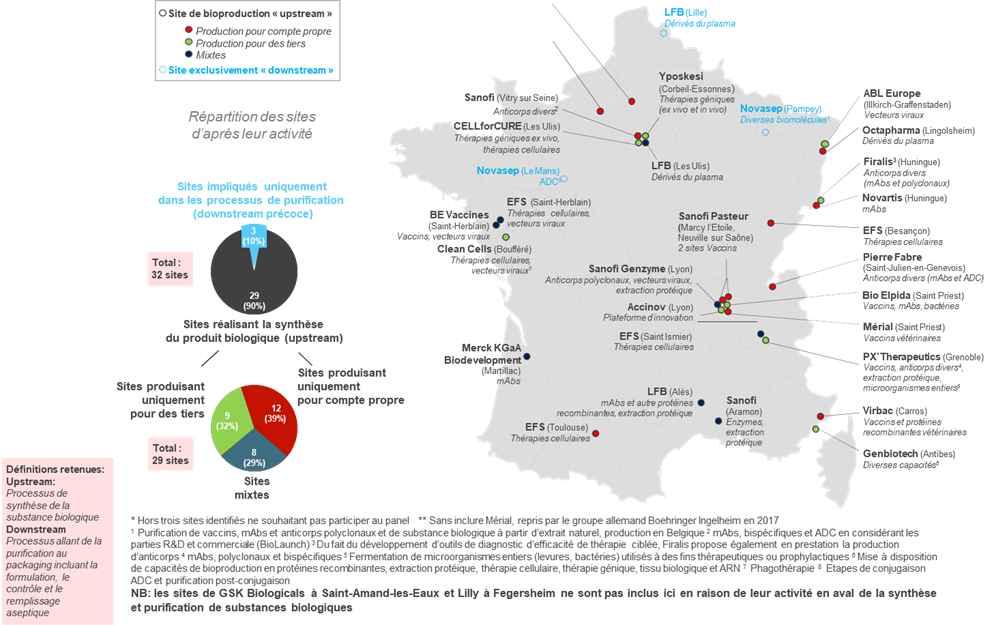
Source : AEC Partners, Cartographie de la bioproduction en France , rapport pour le LEEM, janvier 2018
b) Soutenir la souveraineté sanitaire par la préservation des productions de médicaments matures
Si le virage vers les biotechnologies est une nécessité pour retrouver une place de leader dans le domaine des médicaments innovants, la préservation de la robustesse de l'appareil industriel français ne peut passer par le seul soutien à la filière innovante .
• Les médicaments chimiques matures, dont le brevet a expiré, représentent 49 % des emplois de production en France, soit près de 22 000 emplois.
Les acteurs du secteur pharmaceutique ont déploré une politique budgétaire extrêmement contrainte avec la préoccupation des pouvoirs publics de contenir la progression de l'objectif national des dépenses d'assurance maladie (Ondam). L'industrie pharmaceutique a particulièrement souligné la pression exercée sur l'enveloppe dédiée au médicament au sein de l'Ondam, souvent soumise à un taux de progression limité, de l'ordre de 0,5 % , faisant de l'industrie pharmaceutique une « variable d'ajustement » et lui faisant porter une partie substantielle des efforts de maîtrise de la dépense .
Par ailleurs, la vague d'innovation majeure en cours occasionne des coûts importants mettant sous pression l'enveloppe fermée de prise en charge qui demeure autour de 24 milliards d'euros nets depuis cinq ans. Pour les industriels, le maintien d'une production nationale est en partie menacé par un tel modèle financier.
Le maintien de chaînes de production solides sur les produits chimiques matures est une nécessité pour la structuration de la production pharmaceutique dans son ensemble. En outre, la persistance d'une production de qualité sur le territoire est un atout déterminant dans un secteur en tension régulière sur les approvisionnements - fragilité a été visible durant la crise sanitaire -, mais aussi un outil de prévention des pénuries constatées sur des médicaments anciens qui demeurent essentiels .
Les acteurs de la filière ont fait une série de propositions pour renforcer la souveraineté sanitaire française à travers la consolidation de l'appareil industriel et le renforcement de la compétitivité des entreprises françaises du secteur pharmaceutique. Différents outils financiers ont ainsi été avancés :
- accroître la soutenabilité économique de la filière . L'une des propositions est notamment de mettre au point un dispositif de « prix plancher » dans l'accord-cadre entre le Leem et le Comité économique des produits de santé (CEPS). Il s'agit de déterminer un prix qui intègre les surcoûts occasionnés par une production localisée en Europe ou en France , en assurant à l'exploitant une marge. De tels mécanismes seraient de nature à sécuriser une chaîne de production souveraine , particulièrement pour des médicaments essentiels à la rentabilité fragile, pour des principes actifs ou pour des médicaments d'intérêt thérapeutique majeur faisant l'objet de tensions régulières d'approvisionnement ;
- soutenir l'investissement en reprenant le dispositif de suramortissement mené de 2015 à 2017 ;
- simplifier le cadre réglementaire et financier des TPE-PME.
Proposition n° 2 : soutenir la localisation en France et en Europe de la production pharmaceutique par des mécanismes de prix garantissant des marges aux producteurs et par des dispositifs fiscaux incitatifs.
Proposition n° 3 : assouplir la progression de l'enveloppe « médicaments » au sein de l'Ondam afin de soutenir la production française.
Comme le constate le ministère de la recherche, si aujourd'hui le niveau des prix en France s'est rapproché, pour les produits nouveaux, de la moyenne européenne, et plus encore pour des produits innovants récents, il demeure inférieur au niveau des prix constatés dans les pays avec lesquels la France est en compétition pour la localisation d'activité , notamment l'Allemagne.
• Différentes mesures ont déjà été prises à l'égard de la politique des prix en vue de soutenir la production de médicaments sur le territoire national.
Une action coordonnée du CEPS en soutien
à la production de médicaments :
l'accord-cadre du
5 mars 2021 entre le CEPS et le Leem
Le nouvel accord-cadre prévoit les dispositions suivantes dans son chapitre III, « soutien aux investissements et aux exportations » :
Article 27 : une stabilité du prix facial de cinq ans maximum peut être accordée par le comité à un produit en fonction des investissements en lien direct avec ce même produit dans les capacités de production , la R&D ou des solutions numériques, récemment réalisés ou à venir en Union européenne, notamment en France (y compris par l'intermédiaire d'un façonnier).
L'industriel s'engage à fournir au comité les montants détaillés, investis ou prévus, les quantités effectivement produites pendant la durée de stabilité ainsi que toute information sur les soutiens publics dont il a bénéficié du fait de ses investissements.
Article 28 : hausse de prix lorsqu'une entreprise fait état d'un risque important pouvant impacter la production ou la commercialisation pour une de ses spécialités pharmaceutiques répondant à un besoin thérapeutique qui ne serait plus couvert au cas où elle disparaitrait du marché. Le comité peut, à l'occasion de la demande de hausse de prix d'une entreprise pour un produit qui aurait des concurrents, se saisir pour un motif de santé publique (notamment pour préserver les capacités d'approvisionnement) d'une révision de prix de tout ou partie d'une classe thérapeutique.
Le bénéfice des dispositions prévues au présent article s'accompagne d'un engagement de l'entreprise à approvisionner le marché français. À défaut, s'il est démontré que la responsabilité de l'entreprise est en cause dans la rupture d'approvisionnement, le comité peut aligner le prix facial hors taxe du médicament sur le prix net.
Article 29 : possibilité de bénéficier des avoirs sur remises au titre du guichet du CSIS pour les entreprises éligibles qui ont réalisé des investissements, dans l'Union européenne et notamment en France , visant en particulier le développement des produits, l'augmentation, l'optimisation ou la digitalisation des capacités de production.
Article 30 : le CEPS peut décider d'une stabilité du prix facial pouvant aller jusqu'à une durée de deux ans renouvelable une fois pour deux ans au plus , pour les produits dont au moins une étape significative de fabrication [principe actif, produit fini, conditionnement] est située en Union Européenne, notamment en France, avec libération des lots effectuée en France et dont plus de 60 % des volumes sont exportés.
Source : Réponse de la direction générale de la santé au questionnaire des rapporteures
Cependant, les politiques de soutien à la relocalisation d'une production de médicaments, particulièrement de médicaments innovants, ne doivent pas laisser sur le côté les entreprises aujourd'hui déjà localisées sur le territoire national .
Il convient de valoriser des entreprises ayant historiquement préservé une production localisée en France , notamment de médicaments matures ou de médicaments génériques. Les outils financiers ou fiscaux doivent être également mobilisés pour conserver cette production utile à la chaîne de production et porteuse d'emplois.
2. Mieux accompagner les biotechs françaises dans leur maturation
a) Un « plafond de verre » identifié dans l'accès au financement des biotechs
• Devant la commission des affaires sociales, les représentants de biotechs ont mis en avant un dynamisme du secteur en France, avec la création d'une soixantaine d'entreprises chaque année . Les acteurs s'accordent cependant pour dire que, si le nombre de créations est satisfaisant, il n'est pas un bon indicateur de la vitalité et de la solidité du secteur : ainsi, peu d'entreprises arrivent à mobiliser les fonds nécessaires à des essais de phase 3 et la majeure partie des biotechs peine à franchir un « plafond de verre » .
Les acteurs entendus ont mis en avant un foisonnement de start-ups françaises dans le domaine des biotechnologies, pour beaucoup issues de la recherche académique . Les soutiens financiers publics ont été mis en avant, au premier rang desquels le crédit d'impôt recherche (CIR), le statut de jeune entreprise innovante ou les différents fonds spécifiques.
Dans une année qui a mis sur le devant de la scène des start-ups innovantes qui ont permis une mise en production rapide de vaccins efficaces, l'environnement français de start-ups a été remis en question : pourquoi aucune start-up française n'a émergé en leader dans le domaine ? À cette question, les acteurs s'accordent à répondre que si la France a de très bonnes capacités de pré-amorçage ou d'amorçage, elle peine lourdement à accompagner la maturation des entreprises . Si la France arrive à faire émerger des sociétés d'une valeur de 300-500 millions d'euros, elle n'arrive pas à les faire croître au-delà. À ce titre, le rachat récent d'une start-up de l'Institut national de la santé et de la recherche médicale (Inserm) par le laboratoire Pfizer pour un montant de 800 millions d'euros fait figure d'exception, et le seuil du milliard d'euros n'est jamais franchi.
Le développement des innovations nécessite aujourd'hui des infrastructures et des chaînes d'acteurs complexes, couplées à une grande expertise. Le ministère de la recherche constate que ces changement ont entraîné une complexification des procédés de mise au point des candidats médicaments et une augmentation du coût de développement des produits de santé . À titre d'exemple, le coût moyen de développement d'un médicament commercialisé était estimé à 802 millions de dollars en 2003 ; en 2016, il était de 2 558 millions de dollars.
Ainsi, seulement un ou deux fonds d'investissement semblent en position d'accompagner les start-ups en Europe, et aucun ne serait en capacité de soutenir un investissement supérieur à 500 millions d'euros en France . Les représentants entendus ont tous déploré une vision trop court-termiste de l'investissement et, surtout, une inadéquation de ces investissements avec la catégorie des biotechs , qui portent des innovations de rupture nécessitant une prise de risque de plus long terme . La capacité des acteurs à assumer le risque est donc déterminante , alors qu'on estime ainsi à 2 % les chances de réussite d'une start-up .
Les phases 2 et 3 sont ainsi majoritairement financées par un recours aux marchés financiers, avec un choix fait par les sociétés britanniques et allemandes de se tourner vers les bourses américaines, permettant des levées de fonds plus importantes. Ces blocages conduisent à un horizon pour les start-ups françaises qui demeure souvent le rachat par de grands groupes . Si ce choix peut être favorable pour l'entreprise elle-même, la perspective est négative en termes de création de valeurs et d'emplois en France.
• Sur ce constat de risque pour un secteur stratégique, le secteur des biotechnologies a été intégré en 2020 dans le décret et l'arrêté relatifs au contrôle des investissements étrangers en France 6 ( * ) .
b) Renforcer l'accompagnement financier des développements cliniques des biotechs en consolidant les dispositifs existants
Le besoin de financement pour le développement et la maturation d'une biotech représente un montant considérable, qui peut être de l'ordre de 300 millions d'euros par molécule pour développer un médicament, sur un temps relativement long, de dix à quinze ans . Compte tenu du niveau de risque dans le secteur, le besoin pour une mise sur le marché est souvent estimé autour d'un milliard d'euros.
Face à ces besoins, les acteurs entendus appellent à préserver les fondamentaux du financement français, et particulièrement :
- les acteurs institutionnels comme la Banque publique d'investissement (BPI) reconnue comme un acteur de grande qualité : la création de nouveaux fonds ad hoc ne semble pas nécessaire pour gagner en efficacité ;
- le CIR .
Différentes solutions sont avancées avec notamment, concernant la BPI, un accroissement des investissements en capital-risque .
Une autre recommandation évoquée lors des auditions serait d'adopter un dispositif fiscal incitatif permettant de rediriger de l'ordre de 4 % à 6 % de l'assurance-vie en France vers des fonds d'investissement en innovation, pour un montant estimé à plus de quatre milliards d'euros. Il s'agit notamment de permettre de mettre à disposition sur les marchés français des montants importants permettant de financer les entreprises sans s'orienter nécessairement vers les marchés américains.
Enfin, des leviers européens pourraient être davantage mobilisés avec le lancement d'un projet important d'intérêt européen commun en santé (PIIEC) destiné à accompagner le développement des innovations en santé et d'un appel à manifestation d'intérêt. À l'échelle européenne encore, la future agence HERA aura aussi une mission importante de soutien financier aux biotechs au nom d'un impératif de souveraineté et de sécurité sanitaire au niveau du continent.
• Dans leur ensemble, les acteurs appellent à un changement radical de culture à l'égard du risque . Les stratégies d'investissement dans l'innovation en santé doivent s'orienter plus fortement sur du capital-risque . Cette approche, aussi retenue par les pays anglo-saxons dans leur appréhension de la stratégie vaccinale, permet des paris ambitieux sur des risques finement calculés 7 ( * ) .
Proposition n° 4 : consolider le rôle de la BPI dans le soutien à l'innovation en santé par un accroissement des investissements en capital-risque.
Proposition n° 5 : rendre accessibles sur les marchés français des montants d'investissements majeurs par une meilleure approche du risque et des fonds d'investissements drainant plus fortement les produits d'épargne.
c) Renforcer la dynamique territoriale autour de clusters de taille critique
• La proximité géographique des acteurs est un gage de fluidité dans les échanges et d'accélération de l'innovation. Il est important pour les équipes de recherche d'être proches de l'application clinique comme de la production, afin de favoriser les connaissances mutuelles et les adaptations rapides des innovations en construction . Les acteurs s'accordent unanimement sur la nécessité de favoriser des politiques de sites .
La continuité de la chaîne a souvent été soulignée. À ce titre, les instituts hospitalo-universitaires (IHU) ont été présentés comme un modèle de synergies autour d'une même implantation géographique. Ce type de structures doit être soutenu : il permet des échanges entre chercheurs et ingénieurs avec des passerelles bénéfiques entre la recherche et la partie applicative.
Dans le développement d'une politique de clusters , la cohérence est également un impératif : il ne s'agit pas de remettre à plat la carte des pôles de compétitivité existants mais bien de s'appuyer sur eux et sur l'ensemble des structures permettant aujourd'hui des coopérations fructueuses comme la labellisation « Institut Carnot » ou les agences régionales de l'innovation .
Les représentants des biotechs et des pôles de compétitivité ont mis en avant durant les auditions des exemples de clusters étrangers dont les modèles pourraient être suivis.
Ils ont particulièrement mis en avant le cas de Boston avec les clusters de biotechnologies qui se sont développés avec l'exemple de Kendall Square qui rassemble à la fois de grandes entreprises comme Novartis ou Pfizer, arrivées dans les années 2000, mais aussi un grand nombre de start-ups , le tout à proximité du Massachusetts Institute of Technology , université mondialement reconnue, à Cambridge. C'est dans cette région de Boston qu'est par exemple implantée la biotech Moderna.
En Europe, un modèle d'émergence de clusters de haute performance a été souligné avec les « Spitzencluster-Wettbewerb » . Quatre sont spécialisés en santé, dont le Cluster for Individualized ImmuneIntervention (Ci3) qu'a cofondé BioNTech.
Ils ont souligné dans tous ces exemples l'importance de la continuité de la chaîne de valeur de la recherche fondamentale vers la production des thérapies, avec une proximité géographique des sites de recherche et de production.
La création de clusters doit être une priorité des pouvoirs publics : leur constitution nécessite un financement public important pour la recherche et l'emploi. Il n'est pas question d'en faire un outil au service de l'aménagement du territoire : il s'agit d'être attractif et compétitif . À ce titre, l'émergence d'un cluster d'envergure internationale au sein du Grand Paris paraît une priorité.
Ces clusters doivent, pour être performants, être d'une taille suffisante : sur ce point également, le saupoudrage et l'émiettement ne favorisent pas l'émergence de structures capables de rivaliser avec leurs homologues européens. Les acteurs entendus appellent à ce titre à identifier un ou deux domaines dans lesquels un saut compétitif serait possible et pertinent, et à se concentrer sur ceux-ci .
Dans le domaine pharmaceutique, force est de constater que les secteurs de l'oncologie et de l'immunologie apparaissent comme des priorités évidentes pour lesquelles une structuration est possible pour atteindre une taille critique .
Proposition n° 6 : amplifier la politique de sites en identifiant un ou deux sites prioritaires en France pour constituer des clusters d'envergure internationale soutenus par des financements publics massifs.
B. DONNER À LA RECHERCHE LES MOYENS DE VALORISER SES RÉSULTATS
1. Parfaire la professionnalisation du transfert de la propriété intellectuelle
Si la France a désormais rattrapé son retard dans la création de start-ups en santé, celles-ci nécessitent néanmoins que leur soit garantie la possibilité de détenir, à terme, la propriété intellectuelle de la découverte qu'elles comptent exploiter, que cette découverte soit issue de la recherche publique ou privée. C'est à cette condition que les start-ups seront en capacité d'attirer les investisseurs disposant de la surface financière suffisante pour accompagner leur maturation dans le cadre de développements cliniques de phases 2b et 3 .
a) Faire évoluer, dans certains cas, le mandataire unique en « propriétaire unique »
Déployée à compter de 2014 8 ( * ) , la désignation d'un mandataire unique 9 ( * ) par des personnes publiques pour les brevets détenus en copropriété a été systématisée par la loi « Pacte » de 2019 10 ( * ) et son décret d'application de 2020 11 ( * ) . Plusieurs organismes auditionnés ont toutefois regretté qu'une partie des transferts de propriété intellectuelle, comme la cession des actifs de propriété intellectuelle ou l'engagement contractuel d'une cession ultérieure, échappe encore au périmètre du mandat défini par les copropriétaires 12 ( * ) .
Or ces modalités de transfert sont essentielles pour permettre à des projets à fort potentiel d'attirer des investissements et d'être soutenus financièrement tout au long du développement clinique de l'invention. Pour l'heure, lorsque le mode de valorisation n'entre pas dans le périmètre du mandataire, un comité d'arbitrage est réuni pour acter une décision de principe entre les copropriétaires qui doivent ensuite, chacun, se prononcer sur le contenu de l'accord de cession et formellement le valider. Cette procédure d'arbitrage, longue, ne fait que complexifier et ralentir la conclusion de partenariats entre la recherche publique et des investisseurs privés .
La commission appelle, par conséquent, à parachever la logique de simplification engagée par la mise en place du mandataire unique en renforçant son autonomie dans la conduite des opérations de cession de propriété intellectuelle . Les divergences entre les objectifs de valorisation des copropriétaires ne doivent plus en effet constituer un frein à la conclusion de partenariats avec des acteurs extérieurs. La commission recommande ainsi la transformation progressive du mandataire unique en un véritable « propriétaire unique » dans les cas où cela s'avérerait le plus pertinent, pour plus de lisibilité pour les industriels et les investisseurs privés , en particulier étrangers.
Ce propriétaire unique disposerait d'un mandat élargi et d'une autonomie renforcée pour la négociation et la signature de tous les contrats de transfert, y compris ceux impliquant la cession d'un résultat 13 ( * ) , sous réserve du respect d'une charte de principes partagés et d'un accord sur la répartition des revenus nets , validés par les copropriétaires dès la désignation du mandataire unique afin de ne plus réenclencher de procédure d'arbitrage à chaque opération de cession . Il redistribuerait ensuite entre les copropriétaires les bénéfices tirés de cette valorisation.
Le recours au propriétaire unique est déjà pratiqué dans un certain nombre de pays anglo-saxons afin de permettre aux industriels d'identifier un seul interlocuteur dans leurs négociations de transfert, souhait partagé par un grand nombre de groupes pharmaceutiques internationaux. Il est également pertinent dans des cas où les enjeux de valorisation impliquent, comme le souligne l'institut Pasteur, « de régler des situations précontentieuses, voire d'initier des litiges et négocier des transactions. Ces situations requièrent des réponses agiles et sans délai, incompatibles avec les règles de copropriété. Le principe du mandataire ne permet pas cette agilité, au contraire le mandat exclut les litiges et transactions. » 14 ( * ) L'évolution du dispositif du mandataire unique pourrait ainsi utilement s'inspirer du modèle britannique de valorisation et de transfert articulé autour de l'organisation LifeArc 15 ( * ) .
Proposition n° 7 : ouvrir la possibilité pour le mandataire unique de se transformer en un « propriétaire unique » disposant d'un mandat élargi et d'une autonomie renforcée pour la cession d'actifs et la conclusion de partenariats, sous réserve du respect d'une charte de principes partagés et d'un accord sur la répartition des revenus nets, validés par les copropriétaires dès la désignation du mandataire.
b) Professionnaliser et homogénéiser les pratiques de valorisation des acteurs du transfert
Les critères de transfert de la propriété intellectuelle peuvent sensiblement varier d'une société d'accélération du transfert de technologies (SATT) à une autre. En tenant insuffisamment compte des spécificités des mécanismes de certaines innovations et des besoins des acteurs de l'écosystème territorial, certaines SATT peuvent en effet prévoir des conditions de partage des résultats de la valorisation peu attractives pour les investisseurs 16 ( * ) .
En résulte une demande légitime des acteurs de
la recherche et du développement industriel, notamment au sein des
pôles de compétitivité en santé, de l'engagement
d'une véritable
démarche de
benchmark
des
pratiques de transfert de la propriété intellectuelle en
santé à l'étranger et en France
afin de garantir
une plus grande cohérence des contrats de licence et de gestion de la
propriété intellectuelle. Ce
benchmark
, qui serait
réalisé par une future agence de l'innovation en
santé
- dont les contours sont précisés au
point I. B. 2. du présent rapport -
déboucherait sur un
guide des bonnes
pratiques
de transfert
dans un objectif de professionnalisation des SATT.
Proposition n° 8 : professionnaliser l'activité de valorisation conduite par les SATT dans le domaine de la santé en s'appuyant sur un benchmark des pratiques de transfert qui déboucherait sur un guide des bonnes pratiques pour l'élaboration des contrats de licence.
2. Se donner les moyens d'une recherche ambitieuse en santé
a) Rattraper le retard dans le financement de la recherche publique
La capacité de notre pays à développer des innovations de rupture dans le domaine de la santé est subordonnée au dynamisme de sa recherche fondamentale . Le développement accéléré des vaccins à ARN messager contre la covid-19 masque près de trente ans de recherche fondamentale à laquelle l'Institut Pasteur a pris une part importante.
Or l'évolution des financements publics de la recherche et développement (R&D) en santé en France, qui ont diminué (hors CIR) de 28 % entre 2011 et 2018 17 ( * ) , contraste avec l'augmentation des moyens de la recherche en Allemagne 18 ( * ) et au Royaume-Uni où les crédits publics en R&D en santé ont progressé, respectivement de 11 % et de 16 %. Dans un rapport 19 ( * ) conjoint de mars 2021, les Académies nationales de médecine et de pharmacie font le constat d'un « recul spectaculaire du soutien à la recherche en biologie-santé » en France, avec une diminution évaluée à 25 % entre 2008 et 2020.
Les crédits publics consentis à la biologie-santé ne représentent finalement qu'un peu plus de 17 % du total des moyens de la mission interministérielle de la recherche et de l'enseignement supérieur (Mires) du budget de l'État, soit un niveau sensiblement inférieur à celui observé dans d'autres pays européens - « qui consacrent 35 % à 40 % de leur budget à cette recherche, et jusqu'à 50 % au Royaume-Uni » - et ce, dans un contexte marqué par une multiplication par plus de dix en quinze ans du coût des travaux dans ce domaine.
Budget consacré à la biologie-santé depuis 2006
Données Mires, en millions d'euros constants 2019
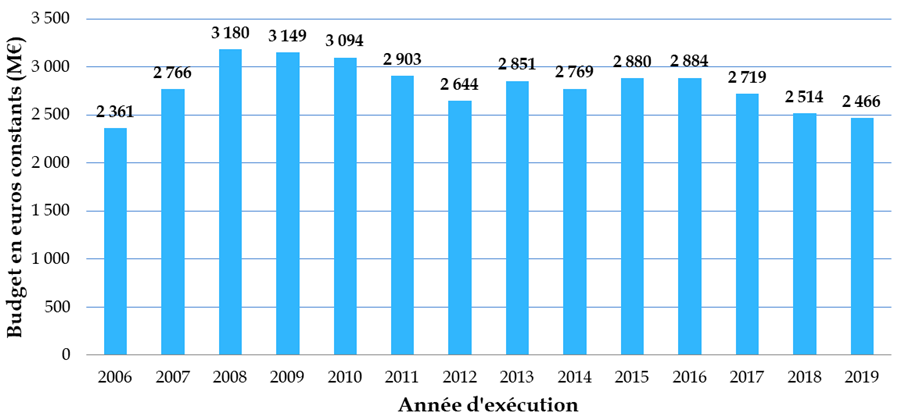
Part des crédits consacrés à la
biologie-santé
dans les moyens de la Mires depuis 2006
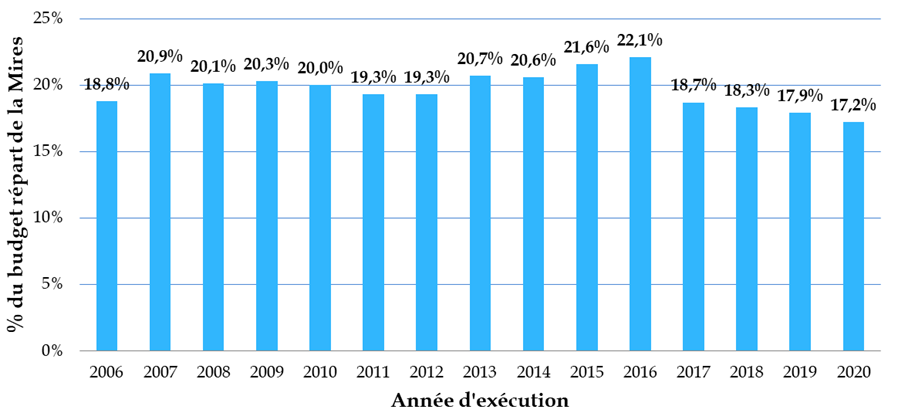
Source : Académie nationale de médecine et académie nationale de pharmacie, Réformer la recherche en sciences biologiques et en santé - Partie I, le financement , rapport bi-académique 30 mars 2021
La loi de programmation de la recherche pour la période 2021-2030 20 ( * ) ne comporte aucun engagement chiffré et pluriannuel concernant l'effort qui sera spécifiquement déployé en faveur de la recherche biomédicale . Encore une fois, la recherche en santé n'est pas clairement identifiée comme une priorité de l'investissement public en recherche, alors que, comme l'indique l'Inserm, la part de la France dans le total des publications scientifiques mondiales s'est érodée de plus d'un tiers de 2005 à 2017 .
Au-delà du sous-financement structurel de la recherche publique , les organismes auditionnés font le constat préoccupant d'un émiettement des sources de financement qui pénalise l'accompagnement de projets à fort potentiel qui nécessiteraient des investissements conséquents pour poursuivre leurs développements. La BPI finance une multitude de projets dans le domaine de la santé mais, en l'absence de vision stratégique, ses soutiens sont insuffisamment hiérarchisés. Quant au PIA 4 21 ( * ) , parmi ses quinze stratégies d'accélération 22 ( * ) , seulement trois concernent à ce stade directement la santé 23 ( * ) .
Dans ces conditions, la commission préconise :
- le doublement de la part des crédits de la mission interministérielle « Recherche et enseignement supérieur » dédiés à la recherche en biologie-santé, afin d'atteindre une proportion de plus de 30 % des financements publics en faveur de la recherche (hors CIR) consentis à la recherche biomédicale ;
- l'identification de quelques secteurs à haut potentiel et stratégiques dans le domaine de la santé et la priorisation pour ces secteurs de l'accès aux financements publics apportés par la BPI et l'Agence nationale de la recherche (ANR). À cet égard, l'action « Programmes et équipements prioritaires de recherche » 24 ( * ) (PEPR) du PIA 4 constituerait un levier pertinent d'accompagnement de ces secteurs prioritaires et d'accélération de leurs développements. Cette priorisation suppose que notre pays assume clairement le choix de se spécialiser dans certains segments de la recherche biomédicale (oncologie, immunologie, cardiologie, maladies infectieuses, biothérapies ou santé mentale...). Ces secteurs prioritaires seraient définis par la future agence de l'innovation en santé, après concertation des acteurs de la recherche académique (organismes nationaux de recherche, alliances scientifiques, universités...), hospitalière et industrielle et des acteurs du système de santé.
Proposition n° 9 :
remédier au sous-financement structurel de la recherche
biomédicale par :
- le doublement de la
part des crédits de la mission interministérielle
« Recherche et enseignement supérieur »
dédiés à la recherche en biologie-santé ;
- l'identification de quelques secteurs à haut potentiel et
stratégiques dans le domaine de la santé en faveur desquels
serait priorisé l'accès aux financements publics.
b) Rénover la gouvernance nationale et européenne de l'innovation en santé
Pendant la crise sanitaire, le rôle des agences américaines, telles que la « Biomedical Advanced Research and Development Authority » (Barda) et la « Defense Advanced Research Projects Agency » (Darpa), a été analysé comme décisif dans la course à l'innovation pour développer des vaccins et des traitements contre la covid-19. Comme l'a rappelé M. Franck Grimaud, président-directeur général de Valneva et président du pôle de compétitivité Atlanpole Biotherapies, la Barda 25 ( * ) est en capacité d'injecter en permanence dix à vingt milliards de dollars dans des projets prioritaires. Dès février 2020, elle a choisi d'investir dans une dizaine de projets de vaccin en soutenant chacun d'eux à hauteur de plusieurs centaines de millions de dollars. Face à une menace d'ampleur, la Barda et la Darpa se sont ainsi clairement positionnées comme des partenaires des développements, en se montrant prêtes à assumer le risque de l'innovation . Par contraste, l'Europe s'est positionnée comme acheteuse auprès des industriels du développement de vaccins, et non comme facteur d'innovation .
L'audition 26 ( * ) par la commission de Mme Kate Bingham, ancienne responsable de la cellule « vaccins » britannique 27 ( * ) , a également mis en lumière la différence d'approche entre l'Europe et le Royaume-Uni dans l'acquisition de vaccins : les négociations conduites par les autorités européennes ont été perçues comme particulièrement complexes et bureaucratiques par nombre d'industriels, quand le pragmatisme du gouvernement britannique, déterminé à accompagner par des financements massifs le développement des candidats les plus prometteurs, a été, lui, unanimement salué.
Tirant les enseignements de la crise sanitaire, la Commission européenne a lancé début 2021 une initiative pour la création d'une autorité européenne de préparation et de réaction en cas d'urgence sanitaire (« European Health Emergency Response Authority » - HERA), chargée d'investir de façon réactive dans le développement et le déploiement de contremesures et l'augmentation des capacités de production 28 ( * ) .
Cette agence, dont la création est annoncée pour 2021, devrait être appelée à coordonner les investissements stratégiques pour la recherche, le développement clinique, la production, le déploiement et la distribution de contremesures . L'enjeu est qu'elle puisse disposer d'une « force de frappe » comparable à celle de la Barda ou de la Darpa, qui se chiffre en plusieurs dizaines de milliards d'euros, afin de financer des projets innovants pour réagir à une menace sanitaire et de partager le risque au niveau européen.
Cette réflexion sur les agences publiques de soutien à l'innovation a relancé le projet de création d'une agence française de l'innovation en santé porté par un certain nombre d'acteurs du secteur, dont France Biotech, tête de réseau des biotechs en France, et le Leem. À l'appui de sa proposition, France Biotech met en avant l'expérience de l' agence de l'innovation de défense 29 ( * ) . Créée en septembre 2018, cette agence constitue un guichet unique pour le dépôt de projets innovants sur une série de thématiques prioritaires.
Lors de son audition par la commission des affaires sociales, M. Franck Mouthon, président de France Biotech, a indiqué voir dans cette agence le chef d'orchestre d'une stratégie de l'innovation en santé , en capacité de « coordonner cette politique d'amont, qui est très bien abordée par la BPI, et cette politique d'aval où on ne parle pas ou très peu et pour laquelle les acteurs ont besoin d'avis engageants, de bénéficier d'éclairages sur la pertinence de leurs développements - question à laquelle le système de santé peut répondre, que ce soit la direction de la sécurité sociale, l'ANSM, la HAS, le Comité économique des produits de santé (CEPS)... » Cette agence pourrait ainsi se voir confier, sur la base de priorités stratégiques clairement définies, le soin de « régler des problèmes sur un engagement de trois mois, sur un transfert de technologie, sur la fixation du prix au niveau du CEPS, sur l'évaluation au niveau de la HAS... »
Convaincue de la nécessité de définir une feuille de route de l'innovation en santé articulée autour de priorités répondant aux besoins de notre système de santé, la commission est favorable à la création d'une agence de l'innovation en santé à la condition que celle-ci ne constitue pas une strate administrative supplémentaire dans un paysage institutionnel de la recherche déjà peu lisible mais qu'elle soit bien un facteur de simplification et d'accélération .
Afin de ne pas créer un nouvel établissement public, l'agence de l'innovation pourrait être constituée, à l'instar de l'agence de l'innovation de défense, sous la forme d'un service à compétence nationale (SCN) placé directement sous l'autorité du ministre de la santé et doté de pouvoirs décisionnels délégués lui ménageant une autonomie d'action sous le contrôle du ministre 30 ( * ) .
Préfiguration de l'agence de l'innovation en santé
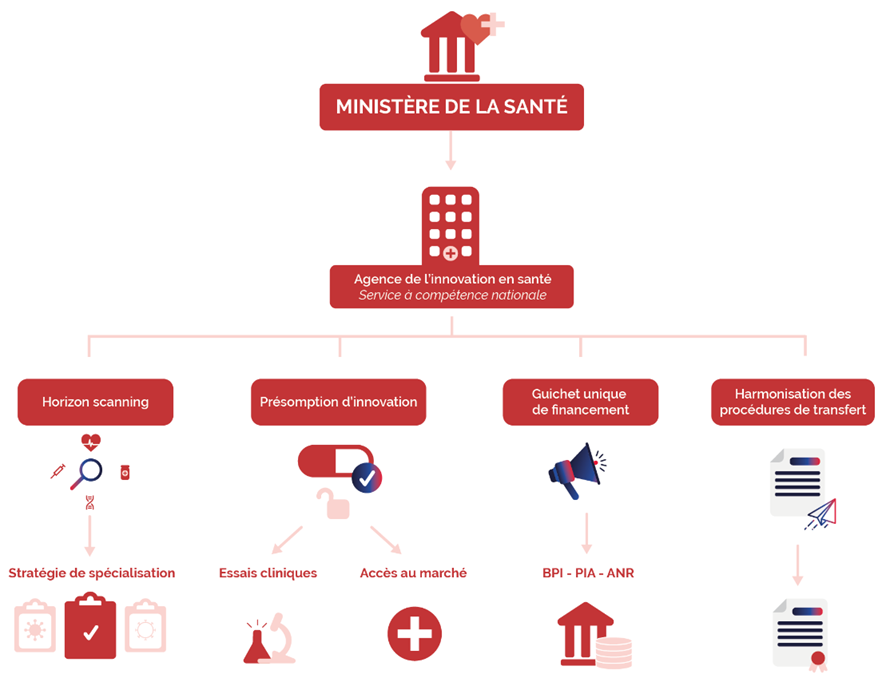
Source : Commission des affaires sociales du Sénat
L'agence de l'innovation en santé serait alors chargée de missions très opérationnelles :
- réaliser une stratégie de veille ( horizon scanning ) des développements de thérapies innovantes les plus prometteurs afin d'élaborer une stratégie de spécialisation sur les segments de recherche porteurs de l'innovation en santé qui devront être soutenus prioritairement. La définition de ces secteurs prioritaires devra s'appuyer sur une consultation large des acteurs de la recherche académique et industrielle et des acteurs du système de santé afin de faire correspondre les développements soutenus à la satisfaction des besoins de notre système de santé ;
- attribuer une présomption d'innovation à certains développements et thérapies s'inscrivant dans les secteurs prioritaires qu'elle aura définis, qui permettra de venir en soutien, le cas échéant, d'une demande d'examen en procédure accélérée (« fast-track ») d'une demande d'autorisation d'essai clinique auprès de l'Agence nationale de sécurité du médicament et des produits de santé (ANSM) et/ou d'un comité de protection des personnes (CPP), d'une demande d'évaluation ou d'accès précoce auprès de la Haute Autorité de santé (HAS) ou d'une négociation de prix auprès du CEPS. L'agence pourra être sollicitée par un promoteur ou un industriel pour obtenir cette présomption d'innovation afin de « débloquer » l'instruction de ce type de demande. La publication des décisions de présomption d'innovation permettra également d'encourager le soutien de ces développements par des investissements privés, notamment par des fonds de capital-risque, qui verront dans la présomption d'innovation accordée par l'agence la garantie d'une mise en oeuvre facilitée des essais cliniques ou d'un accès accéléré au marché ;
- constituer un guichet unique pour le dépôt de dossiers de candidature à l'ensemble des appels d'offres pour l'obtention de financements publics de la recherche en santé, afin de flécher des investissements publics massifs sur des développements en phases précliniques ou cliniques . À ce titre, l'agence centralisera pour le compte de la BPI, du SGPI 31 ( * ) , de l'ANR et du MESRI 32 ( * ) les candidatures aux appels d'offres organisés par ces structures et permettra le dépôt d'un seul et même dossier pour plusieurs appels d'offres en lien avec l'innovation en santé, afin d'alléger la charge administrative des équipes de recherche ;
- simplifier et harmoniser les procédures de transfert de propriété intellectuelle par l'élaboration de guides de bonnes pratiques.
Proposition n° 10
: créer une agence de l'innovation en santé sous la forme d'un
service à compétence nationale placé sous
l'autorité du ministre de la santé, chargée des missions
suivantes :
- définir, à partir d'un
horizon
scanning
, une stratégie de spécialisation sur les segments
de recherche porteurs de l'innovation en santé qui devront être
soutenus prioritairement pour répondre aux besoins du système de
santé ;
- attribuer une
présomption d'innovation à des développements et
thérapies s'inscrivant dans ces segments prioritaires afin de faciliter
le lancement d'un essai clinique ou un accès rapide au
marché ;
- constituer un guichet
unique pour le dépôt centralisé d'un seul et même
dossier de candidature aux appels d'offres en faveur de l'innovation en
santé ;
- simplifier et
harmoniser les procédures de transfert de propriété
intellectuelle par l'élaboration d'un guide de bonnes
pratiques.
II. DÉVELOPPER L'ACCÈS DES PATIENTS À L'INNOVATION EN PRENANT RÉSOLUMENT LE VIRAGE DE LA MÉDECINE PERSONNALISÉE
A. PERMETTRE À LA FRANCE DE RETROUVER SON ATTRACTIVITÉ POUR ACCUEILLIR LA RECHERCHE CLINIQUE
1. Moderniser l'examen des demandes d'essais cliniques
a) Un bilan en demi-teinte du raccourcissement des délais d'autorisation
Le 8 e CSIS comportait un engagement sur la réduction des délais d'autorisation des essais cliniques respectivement à 60 jours au niveau des CPP , 45 jours pour les médicaments et les dispositifs médicaux (DM) et dispositifs médicaux de diagnostic in vitro (DMDIV) et 110 jours pour les médicaments de thérapie innovante (MTI) au niveau de l'ANSM.
Cet engagement a été globalement respecté par l'ANSM, puisqu'en 2019, le délai moyen de décision sur tous les essais 33 ( * ) s'est établi à 46,5 jours. Ce délai s'est légèrement dégradé en 2020, à 51 jours, en raison de la charge de travail consécutive à la crise sanitaire. Toutefois, grâce à une priorisation et à la mise en oeuvre de procédures accélérées, sur 105 essais cliniques portant sur la covid-19, la durée moyenne de décision a été de 26 jours.
Les actions d'amélioration des délais d'autorisation mises en oeuvre par l'ANSM
L'ANSM a mis en place deux dispositifs de traitement rapide (« fast-track ») qui ont permis de réduire drastiquement les délais d'autorisation des essais cliniques répondant à des critères de priorité de santé publique, avec des délais inférieurs aux exigences règlementaires :
- le dispositif « fast-track 1 » - accès à l'innovation - permet un accès rapide pour les patients aux traitements innovants dans les essais (autorisation en 40 jours au maximum). Les critères d'éligibilité à ce dispositif sont les suivants : essais précoces en oncopédiatrie et hémato-pédiatrie, maladie rare, design complexe, MTI, soutien à l'innovation (c'est-à-dire suite d'une recherche du programme hospitalier de recherche clinique - PHRC, d'un centre labellisé de phase précoce - CLIP...) ;
- le dispositif « fast-track 2 » - soutien au développement - permet d'accélérer la mise en place des essais cliniques pour les molécules ou les associations de molécules déjà évaluées par l'ANSM (autorisation en 20 jours maximum). Les critères d'éligibilité à ce dispositif sont les suivants : molécule ou association de molécules déjà évaluées en France et dans la même indication que l'essai concerné, MTI.
Source : Agence nationale de sécurité du médicament et des produits de santé
Les progrès dans l'instruction des demandes d'essais cliniques par les CPP restent plus mesurés. Pour mémoire, les délais règlementaires pour les avis des CPP à compter de la date de recevabilité du dossier s'établissent à 45 jours pour des dossiers ne suscitant pas de questions du CPP et à 60 jours pour les dossiers pour lesquels le CPP adresse des demandes d'informations complémentaires au promoteur. Selon les données de la direction générale de la santé, le délai médian global des essais cliniques de médicaments, tous domaines confondus, s'établit à 77 jours sur les trois premiers mois de 2021. Néanmoins ce délai est calculé à partir de la date du tirage au sort pour la désignation du CPP, et non pas à compter de la date de réception du dossier complet conditionnant sa recevabilité, et comprend également le délai de réponse et de mise en conformité applicable au promoteur.
La conférence nationale des CPP (CNCPP) estime que le temps médian d'évaluation des CPP pourrait s'établir à 53 jours en retranchant le temps de réponse des promoteurs. En décomptant également de ce délai une dizaine de jours pour rendre le dossier complet et recevable, la CNPP considère que le délai médian serait ramené à un niveau proche du délai règlementaire de 45 jours pour les dossiers ne faisant pas l'objet de questions. Néanmoins, concernant plus spécifiquement les recherches en cancérologie, la CNCPP constate une dégradation du délai médian global d'évaluation à 79 jours au premier trimestre 2021, alors que le délai règlementaire d'évaluation prévoit une décision du CPP en 45 jours. À cela s'ajoute une augmentation du délai de réponse des promoteurs qui aggrave le délai global d'autorisation des essais.
Les actions d'amélioration des délais d'instruction des CPP
Un décret du 19 mars 2021 34 ( * ) est venu simplifier le fonctionnement des CPP :
- il ouvre la possibilité d'organiser des réunions par conférence téléphonique ou audiovisuelle pour tout ou partie des membres ;
- il tire les conséquences de la loi de 2018 sur la modulation du tirage au sort des CPP : le système d'information mis en place par la DGS ne met en concurrence que les CPP disponibles et disposant de la compétence nécessaire à l'examen du projet, ces critères devant être précisés par un arrêté. Lorsque le CPP désigné estime qu'il n'est pas en capacité de traiter le dossier, il peut demander au ministre de la santé dans un délai de deux jours de le renvoyer vers un autre CPP ;
- il impose au promoteur un délai maximal de douze jours pour répondre aux questions du CPP. Avec l'entrée en vigueur de cette disposition, à ce jour, sur 348 dossiers, 232 demandes ont dépassé le délai de douze jours et ont donc été réputées caduques.
À la suite d'un audit, la direction générale de la santé a lancé un plan « ambition CPP 2019-2022 » qui contribue à la formation des membres des CPP à l'utilisation du nouveau système d'information SIRIPH2G et à l'application des règlements européens sur les dispositifs médicaux. 11 CPP volontaires vont également être formés spécifiquement pour être les pilotes dans l'application de la procédure européenne d'évaluation des demandes pour les essais cliniques de médicaments selon le règlement européen. L'objectif est, par ailleurs, de doter chaque CPP d'1,5 équivalent temps plein (ETP) permanent, soit 58,5 ETP au total. Les CPP ont obtenu 850 000 euros supplémentaires pour le financement de 17 ETP d'assistants de recherche clinique, mais seuls cinq ou six ETP ont jusqu'ici été effectivement recrutés compte tenu des procédures de recrutement complexes au sein des hôpitaux. En outre, la loi de financement de la sécurité sociale pour 2021 35 ( * ) a procédé à une augmentation de la contribution sur le chiffre d'affaires des industriels pharmaceutiques dont le rendement supplémentaire doit être affecté au financement des CPP.
Source : Conférence nationale des comités de protection des personnes
b) Des essais cliniques précoces insuffisamment accueillis en France
L'accueil d'essais cliniques est un enjeu concurrentiel majeur. La mise en oeuvre en France d'essais précoces est déterminante pour inciter un industriel à poursuivre le développement clinique et la production d'une thérapie innovante sur notre territoire.
Or, en dépit de progrès observés dans la période récente, la France peine encore à se positionner en première ligne pour attirer des essais de phase précoce. Selon les données du Leem 36 ( * ) , la France se situait, au premier semestre 2019, au 4 e rang européen en participant à 12,9 % des essais mondiaux, derrière l'Allemagne (16 %) et le Royaume-Uni et l'Espagne (14,5 % chacun), mais occupait le 5 e rang européen pour l'accueil d'essais de phase 1 en n'accueillant que 5 % de ces essais.
L'ANSM met toutefois en avant une augmentation du nombre d'essais précoces autorisés en France au cours des dernières années, même si notre pays reste sérieusement concurrencé par d'autres pays européens. De 2018 à 2020, la part de la France dans les autorisations d'essais précoces innovants en oncologie s'établit à 21 %, soit un niveau supérieur à l'Allemagne (18 %) et la Belgique (13 %), mais derrière l'Espagne (26 %) et le Royaume-Uni (22 %).
Demandes d'autorisation d'essai clinique enregistrées par l'ANSM
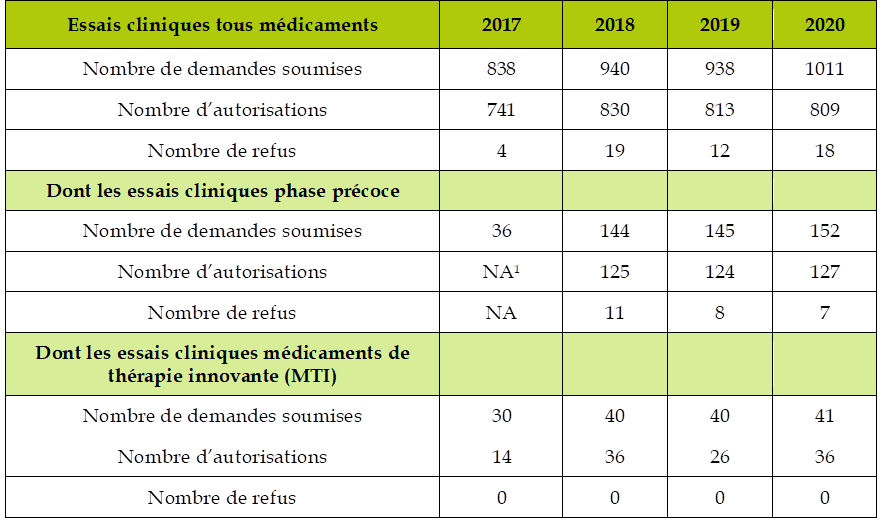
Source : Agence nationale de sécurité du médicament et des produits de santé
Certains groupes pharmaceutiques privilégient clairement d'autres pays que la France pour l'accueil d'essais cliniques. Aucun essai de phase 1 ou 2 n'a ainsi été lancé en France par la biotech française Valneva. Son PDG, M. Franck Grimaud, regrette une attitude très théorique et insuffisamment pragmatique des instances responsables de l'instruction des demandes d'essais cliniques dans l'évaluation des bénéfices-risques. Lors de son audition par la commission 37 ( * ) , il a pointé un problème culturel en France dans l'approche des demandes d'essais cliniques : « l'innovation dans le domaine des biotechnologies repose forcément sur de nouvelles approches, de nouveaux types de médicament, et emprunte souvent un nouveau mode de “ delivery ”. Par essence, la plupart des innovations en biotechnologie sortent des cases, de ce qui est déjà connu. Or, en France mais pas seulement, on privilégie une approche “ to the book ” : si on ne rentre pas dans les cases, dans des choses déjà décrites, on demande un niveau de protection maximal, alors que d'autres pays adoptent une approche plus pragmatique du rapport bénéfices-risques. »
c) Une expertise parfois critiquée
Nombreux sont en effet les promoteurs qui déplorent une certaine frilosité de la part des experts mobilisés par l'ANSM ou les CPP qui manqueraient de pragmatisme dans l'évaluation du rapport bénéfices-risques en suivant une approche essentiellement théorique et monodimensionnelle. Cette frilosité résulterait :
- d'une insuffisante connaissance des nouveaux mécanismes des innovations de rupture qui peut conduire les experts à faire preuve d'une aversion disproportionnée au risque ;
- des contraintes liées à la prévention des conflits d'intérêts qui disqualifieraient nombre d'experts disposant d'une expérience au sein de l'industrie. Cette conception rigide ne ferait qu'aggraver l'éloignement des experts « officiels » de l'ANSM et des CPP vis-à-vis de l'expertise développée dans les territoires qui se nourrit des interactions entre la recherche académique et l'industrie.
Dans un article de 2018 38 ( * ) , le professeur Didier Truchet souligne qu'« on a trop tendance en pratique à confondre liens d'intérêts et conflits d'intérêts, alors pourtant que les textes font la distinction », en rappelant qu'un lien d'intérêts, qui doit être déclaré, « peut conduire à écarter l'expert ou ce dernier à se déporter, mais sous cette réserve, ne le disqualifie normalement pas, en particulier en présence d'une pluralité d'experts ».
À cet égard, la commission plaide pour un renforcement des capacités de la commission nationale des recherches impliquant la personne humaine (CNRIPH) afin de constituer un annuaire des experts mobilisables dans différentes aires thérapeutiques à la disposition des CPP et de mener un benchmark de la gestion des liens d'intérêts dans les autres pays pour en tirer un guide des bonnes pratiques . Elle préconise également la mise en place d'un déontologue de l'expertise sanitaire au sein de la CNRIPH qui permettrait de conseiller les CPP dans leur recours aux experts, dans un souci de préservation des principes d'impartialité, de transparence, de pluralité et du contradictoire de l'expertise sanitaire 39 ( * ) .
d) Des CPP embolisés par l'examen des études observationnelles
Le nombre de recherches examinées par les CPP augmente en moyenne de 8 % par an. En dix ans, le nombre de demandes d'autorisation de recherches impliquant la personne humaine (RIPH) a doublé, pour atteindre 3 987 demandes initiales en 2020 . Cette augmentation est en grande partie alimentée par les demandes portant sur les RIPH de catégorie 3, correspondant aux études observationnelles. Globalement, plus de 35 % des dossiers examinés par les CPP en 2020 étaient des RIPH de catégorie 3, contre 26 % de recherches sur les médicaments et 12 % de recherches sur les dispositifs médicaux.
Répartition des demandes d'autorisation de RIPH par catégories
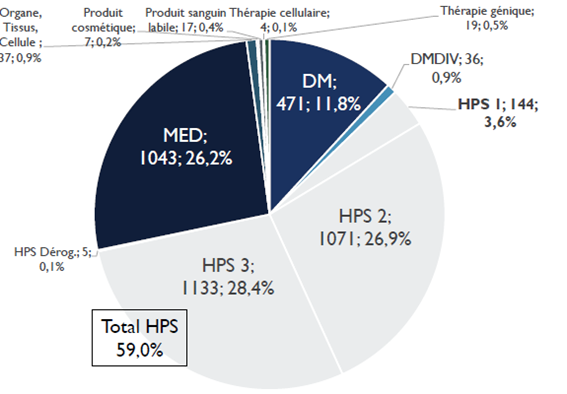
Source : Conférence nationale des comités de protection des personnes
Nombre de RIPH toutes catégories confondues
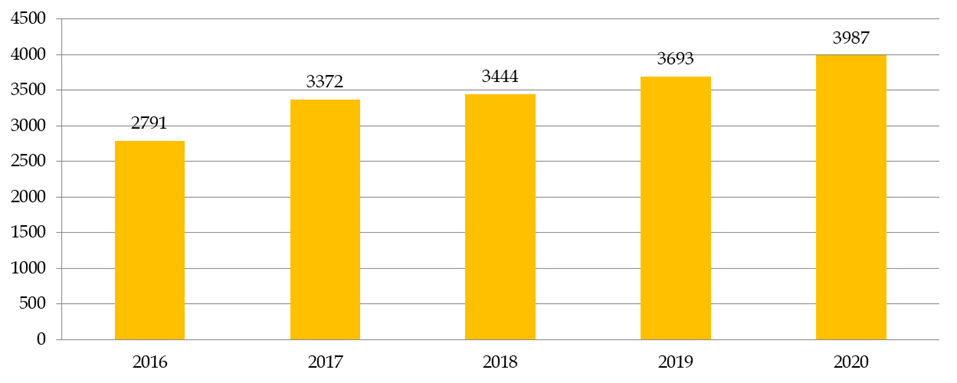
Source : Commission des affaires sociales du Sénat, d'après la direction générale de la santé
Les principales aires thérapeutiques concernées par les RIPH
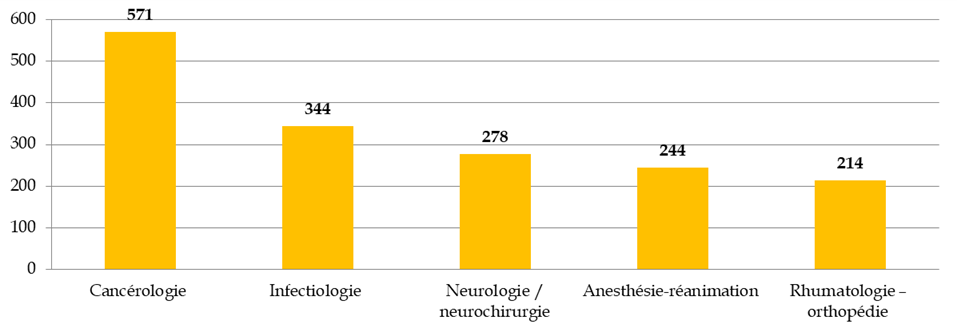
Source : Commission des affaires sociales du Sénat, d'après la direction générale de la santé
Les CPP, au nombre de 39 sur le territoire, ne disposent pas des moyens humains, financiers et techniques pour absorber une telle charge de travail. La loi d'accélération et de simplification de l'action publique 40 ( * ) a, certes, simplifié les procédures applicables aux recherches non interventionnelles ne portant pas sur des produits de santé (HPS) auprès des CPP 41 ( * ) . Cette mesure, encore insuffisamment connue des promoteurs, peine à produire ses effets. Elle ne comprend, du reste, pas d'automaticité pour l'obtention d'un avis favorable du CPP sur des protocoles dont la méthodologie a pourtant déjà été validée par la commission nationale de l'informatique et des libertés (CNIL).
La proportion des RIPH de catégorie 3 a, en outre, vocation à augmenter, les fabricants de dispositifs médicaux étant appelés à réaliser des recherches post-commercialisation. La commission estime donc indispensable d'alléger la charge de travail des CPP pour leur permettre de se concentrer sur l'examen des projets de recherches interventionnelles, en transférant l'examen des RIPH de catégorie 3 à un seul comité d'éthique qui serait spécialisé sur les recherches non interventionnelles et la protection des données de santé personnelles, ainsi que le préconise la présidente de la commission des affaires sociales, Mme Catherine Deroche, dans une proposition de loi déposée en 2019 42 ( * ) .
Outre cette mesure, la commission appelle également à moderniser le fonctionnement des CPP en mettant en oeuvre les mesures déclinées dans la proposition ci-dessous.
Proposition n° 11
: moderniser le fonctionnement des CPP par :
- le transfert de
l'examen des RIPH de catégorie 3 à un comité
d'éthique spécialisé sur les recherches non
interventionnelles et la protection des données de santé
personnelles ;
- le renforcement de la
formation de leurs membres aux mécanismes des innovations de
rupture ;
- la constitution, au plus tard avant la fin 2021,
sous l'égide de la CNRIPH, d'un annuaire d'experts selon
différentes aires thérapeutiques à la disposition des CPP,
avec publication de leurs déclarations d'intérêts ;
- l'assouplissement, sous le contrôle de la
CNRIPH qui serait dotée d'un déontologue de l'expertise
sanitaire, des conditions de mobilisation de l'expertise pertinente en
s'appuyant, à cet égard, sur un
benchmark
des pratiques
dans d'autres pays européens en matière de prévention des
conflits d'intérêts ;
- la rénovation
des modes d'indemnisation des membres et experts des CPP pour valoriser leur
engagement, notamment en reconnaissant l'investissement des présidents
et vice-présidents
43
(
*
)
;
- la mise en place d'un
audit indépendant et périodique des CPP conditionnant le
renouvellement de leur agrément.
La commission appelle également à clarifier la situation des études portant sur la personne humaine mais n'ayant pas de finalité biologique ou médicale , dites « études hors loi Jardé 44 ( * ) ». Ces recherches, qui nourrissent pour partie les sciences humaines et sociales, restent l' angle mort de l'encadrement de la recherche sur la personne humaine , alors que certaines d'entre elles peuvent présenter des risques éthiques pour les personnes 45 ( * ) . À l'heure actuelle, ces recherches sont examinées par des comités d'éthique de la recherche (CER) qui ne font l'objet d'aucun encadrement juridique et dont les pratiques restent très hétérogènes. En outre, par souci de sécurisation juridique, les études impliquant la collecte de nouvelles données et supposant le consentement des personnes à l'utilisation de ces données empruntent bien souvent la voie des RIPH de catégorie 3 et sont alors envoyées pour avis aux CPP, ce qui tend à augmenter d'autant la charge de travail de ces derniers.
Proposition n° 12 : inscrire dans la loi un statut des comités d'éthique de la recherche chargés d'examiner les protocoles de recherche n'ayant pas de finalité biologique et médicale et clarifier les méthodologies de référence applicables à ces recherches.
e) Prendre le virage des essais adaptatifs et des études post-commercialisation
La commission insiste sur l'importance du développement des essais adaptatifs et des études post-commercialisation , après obtention de l'autorisation de mise sur le marché (AMM). Les essais cliniques traditionnels, par définition, procèdent à une évaluation de l'efficacité d'un médicament ou d'une thérapie selon plusieurs biais, notamment les indications visées ou les caractéristiques des patients recrutés, et ne prévoient pas la possibilité de modifier le protocole en cours de mise en oeuvre.
Or, dans une approche de l' innovation au service de l'amélioration continue des soins , la France doit prendre le virage des essais adaptatifs et de l'évaluation en vie réelle. La crise sanitaire a été un facteur d'accélération dans la diffusion des méthodologies adaptatives : les essais « Recovery », au Royaume-Uni, et « Discovery », en France, ont prévu la possibilité d'abandonner en temps réel des traitements expérimentaux apparus inefficaces et de les remplacer, le cas échéant, par d'autres thérapies en fonction de l'évolution des connaissances.
La collecte de données en vie réelle , après la commercialisation d'un produit, permet, en outre, de disposer de données plus robustes pour objectiver l'apport d'un médicament dans la prise en charge des patients mais aussi pour l'efficience du système de santé, en contribuant à l'analyse médico-économique.
Proposition n° 13 : prendre le virage des essais adaptatifs, des études post-AMM et de la collecte de données en vie réelle dans un objectif d'amélioration continue de la qualité des soins.
Une partie significative des médicaments innovants ou des nouvelles indications thérapeutiques pour des médicaments existants résultera, à l'avenir, de la mise en corrélation d'une grande variété de données sur les effets d'une molécule. L'accès aux données du Health Data Hub , qui devrait permettre de disposer d'une multitude de points de mesure, représente une opportunité de réduction des coûts non négligeable pour la conception et l'organisation d'essais cliniques.
La méthodologie de référence MR004 établie par la CNIL, ne requérant qu'un simple engagement de conformité au référentiel, a d'ailleurs permis de faciliter la réutilisation de données issues d'une recherche antérieure ou du soin dans le cadre de recherches translationnelles ou du partage de données avec un chercheur académique ou un industriel. En revanche, la fédération nationale des centres de lutte contre le cancer Unicancer a relevé une lourdeur administrative dans l'appariement de ces données avec celles du Health Data Hub , dès lors que la procédure requiert une autorisation de la CNIL après avis favorable du comité éthique et scientifique pour les recherches, les études et les évaluations dans le domaine de la santé (Cesrees).
Pour mémoire, l' article 18 du projet de loi relatif à la bioéthique permet, par dérogation à l'exigence d'un consentement exprès et préalable, de réutiliser des échantillons humains prélevés initialement à d'autres fins afin de réaliser des examens génétiques à la condition que la personne concernée ait été dûment informée de l'utilisation potentielle de ses échantillons dans le cadre d'un programme de recherche 46 ( * ) , et qu'elle n'a pas exprimé son opposition. Ce mécanisme pourrait utilement être étendu à la réutilisation des données collectées à l'occasion de soins ou d'essais cliniques : le patient serait alors informé de la possibilité de s'opposer à la réutilisation de ses données dans le cadre de recherches s'inscrivant dans un programme de recherche dans le domaine thérapeutique concerné.
Proposition n° 14 : mettre en place une procédure accélérée ( fast-track ) pour permettre l'accès des chercheurs et des promoteurs et investigateurs d'essais cliniques aux données de santé du Health Data Hub pour évaluer l'ensemble des effets d'une thérapeutique.
Proposition n° 15 : étudier la faisabilité de la transposition du dispositif de passerelles soin/recherches de l'article 18 du projet de loi relatif à la bioéthique à la réutilisation de données de santé dans le cadre de programmes de recherche.
Enfin, la crise sanitaire a montré que l'accès aux données de santé constitue une véritable carte à jouer pour attirer des essais cliniques . En témoignent la mise en place au Royaume-Uni d'un registre national de volontaires pour participer à des essais cliniques sur des vaccins ou des traitements contre la covid-19 ou encore l'accès du laboratoire Pfizer aux données de santé des personnes vaccinées en Israël.
La commission appelle à saisir cette opportunité afin d'attirer des essais cliniques, notamment de phase précoce, en mettant en place des registres nationaux de volontaires sur de grands enjeux thérapeutiques (cancers à pronostic sombre, VIH, maladies infectieuses ne disposant pas de traitement ou de vaccin comme Zika, la dengue ou Lyme...).
Proposition n° 16 : créer des registres nationaux de candidats à des essais cliniques sur de grands enjeux thérapeutiques.
2. Rénover le modèle de financement de la recherche clinique
Les missions d'enseignement, de recherche, de référence et d'innovation 47 ( * ) (Merri), censées rétribuer l'investissement des hôpitaux dans la recherche, ne sont en réalité pas vues par ces derniers comme une dotation propre dédiée à leur activité de recherche mais plutôt comme une compensation de la perte de recettes liée au fait que leurs équipes se consacrent à des projets de recherche et non à des activités rémunérées dans le cadre de la tarification à l'activité (T2A).
À cela s'ajoute une approche du financement de la recherche à l'hôpital qui reste centrée sur le volume de publications scientifiques, et apparaît insuffisamment tournée vers la valeur ajoutée de la recherche clinique, qui devrait être appréciée à l'aune des applications industrielles potentielles ou des sauts thérapeutiques ou organisationnels poursuivis. Bien qu'il ait été rénové en avril 2021, le modèle de fixation de la dotation des Merri est ainsi articulé autour de trois enveloppes : l'enveloppe « publications » (61 % de la dotation), l'enveloppe « recherches-inclusions » (15 %) et l'enveloppe « enseignement » (24 %) 48 ( * ) . En conséquence, une part non négligeable de la recherche clinique hospitalière est uniquement accompagnée par les appels à projets et non par les crédits récurrents des Merri.
La commission recommande, dès lors, de revaloriser l'enveloppe « recherches-inclusions » au sein des Merri à hauteur de 25 % de la dotation socle - soit une augmentation de dix points. Cette enveloppe doit également permettre de soutenir les projets d'innovation organisationnelle, notamment en télémédecine et en télésurveillance.
Proposition n° 17 : augmenter l'enveloppe « recherches-inclusions » au sein des Merri à hauteur de 25 % de la dotation socle.
Lors de son audition par la commission, M. Franck Mouthon, président de France Biotech, s'est félicité des progrès réalisés dans la généralisation de la convention unique , mise en place à la suite du CSIS de 2013, qui permet de facturer à l'industriel, dans le cadre d'un seul contrat, les surcoûts engendrés pour les établissements de santé dans l'organisation d'un essai clinique. Il a néanmoins relevé une faiblesse dans l' établissement de l'annexe financière de la convention qui requiert bien souvent une longue négociation avec plusieurs établissements.
Selon des données du Leem, le délai médian pour la conclusion de la convention unique s'est établi à 76 jours en 2018 et 70 jours en 2019, un délai qui s'ajoute aux délais d'autorisation des essais par l'ANSM et les CPP : en moyenne, 204 jours sont ainsi nécessaires en France pour inclure le premier patient depuis le dépôt de la demande d'essai auprès de l'ANSM, contre un objectif cible de 120 jours . La part des essais ayant franchi l'étape de la convention unique dans des délais inférieurs à ce qui serait nécessaire pour répondre à l'objectif cible a stagné à 21 % en 2019, alors que la part des essais ayant franchi l'étape de l'ANSM et celle des CPP dans des délais infra-règlementaires a plus nettement progressé 49 ( * ) .
Proposition n° 18 : confier aux fédérations d'établissements de santé et aux organisations représentatives des promoteurs le soin d'établir des référentiels de coûts et de surcoûts et des méthodologies de calcul afin d'accélérer la négociation de l'annexe financière de la convention unique.
Enfin, la commission plaide pour une simplification du bénéfice du CIR pour couvrir l'ensemble des études et démarches associées au lancement d'un essai clinique . À l'heure actuelle, seules les étapes de conception et de méthodologie des essais cliniques, et d'analyse et de publications ouvrent droit au CIR. L'extension du CIR aux étapes de faisabilité et de mise en place d'un essai clinique pourrait ainsi constituer un facteur puissant d'attractivité .
Proposition n° 19 : étendre l'éligibilité au CIR à l'ensemble des étapes de faisabilité (démarches pour trouver des patients, évaluation de la disponibilité des centres, monitoring ...) et de mise en place des essais cliniques (choix du pays où se dérouleront les essais, démarches règlementaires et d'éthique, formation des personnels des centres...).
B. REPLACER LE PATIENT AU CoeUR DE L'INNOVATION
1. Lever les freins dans l'accès précoce aux innovations
a) Des délais d'accès encore trop longs
• Une préoccupation forte des acteurs du secteur est de permettre un accès rapide des patients à l'innovation . En matière pharmaceutique, l'Union européenne 50 ( * ) a fixé à 180 jours le délai de référence pour l'accès au marché . Ce délai correspond à la durée considérée comme le maximum pouvant séparer l'autorisation de mise sur le marché et la prise en charge du médicament.
La loi reprend ce délai d'accès, fixé à 180 jours pour la ville comme pour la liste en sus, et à 75 jours dans le cas d'une rétrocession. Le respect de ces délais était l'un des engagements du Gouvernement à l'issue du 8 e CSIS.
Le ministère de la recherche a souligné sur ce sujet ce qu'il considère comme « un problème structurel majeur » : les autorisations de mise sur le marché (AMM) des médicaments sont attribuées par l'Agence européenne du médicament (« European Medicines Agency » - EMA), permettant la commercialisation sur tout le territoire européen sans frontières internes, dans le cadre du marché commun, alors que la fixation des prix des médicaments est une compétence étatique , amenant à des disparités intra-européennes en termes de prix et de réglementations. Dans les faits, comme le précise le ministère, il peut exister en France un délai de plusieurs années entre l'autorisation de mise sur le marché obtenu à l'EMA et l'autorisation de vente du produit qui dépend des négociations au CEPS.
Les acteurs du secteur pharmaceutique ont tous regretté des délais encore très longs d'accès au médicament, même si la situation tend à s'améliorer sur les dernières années.
• Selon le CEPS 51 ( * ) , en moyenne, le délai de 180 jours a été respecté en 2019 pour les inscriptions en ville et sur la liste en sus, avec respectivement 144 et 147 jours. Cependant, ces délais moyens sont plus faibles pour les génériques et biosimilaires et plus longs pour les produits issus des ATU et post-ATU.
Derrière ces délais moyens, de grandes disparités apparaissent, avec des délais beaucoup plus longs en ce qui concerne les médicaments innovants.
Ainsi, selon les études réalisées par le Leem 52 ( * ) :
- le délai moyen d'évaluation par la HAS était de 168 jours en moyenne en 2020 selon la base de données Prioritis ;
- le délai moyen de négociation de prix et de publication au Journal officiel était de 229 jours pour les nouvelles spécialités (hors génériques et biosimilaires) en 2019 (dont 62 jours de publication au Journal officiel ).
Concernant les nouveaux médicaments ayant reçu une première AMM, l'étude européenne réalisée par les industries pharmaceutiques estime à 527 jours en 2020 le délai moyen d'accès en France 53 ( * ) . Il est très difficile d'estimer finement le délai d'accès selon l'amélioration du service médical rendu (ASMR) et le délai réel d'accès pour les médicaments innovants en primo-inscription. Le Leem propose une meilleure identification, dans ces délais moyens, des médicaments innovants, notamment ceux bénéficiant d'une présomption d'innovation reconnue par l'Agence européenne des médicaments, mais aussi ceux bénéficiant d'une autorisation temporaire d'utilisation (ATU) : considérer l'ATU comme un délai nul ferait chuter le délai moyen à 257 jours .
Selon les industriels, la France obtient la 21 e place sur 28 pays en termes de rapidité d'accès aux molécules avec un délai moyen de disponibilité de la molécule pour le patient français de 15 à 18 mois.
• La HAS a présenté en janvier 2020 un plan d'action pour les médicaments innovants, autour de trois axes :
- rendre des avis conditionnels, le temps de lever les incertitudes ;
- suivre les médicaments en vie réelle pour vérifier les promesses initiales ;
- renforcer l'agilité de la HAS pour mieux accompagner l'innovation. Sur ce dernier point, la HAS entend principalement se concentrer sur des évaluations à forte valeur ajoutée mais aussi promouvoir les procédures d'évaluation accélérées (dites de « fast-track »).
Le bilan intermédiaire de la HAS est jugé positif avec une montée en charge du dispositif d'évaluation anticipée qui a montré des résultats sur les délais avec un délai moyen de traitement passé de 126 à 105 jours entre 2019 et 2020 pour une proportion de dossiers ayant une évaluation complète qui a augmenté pour atteindre 68 % en 2020. Concernant la délivrance d'évaluations conditionnelles pour certains médicaments prometteurs dans des maladies graves en situation de besoin médical non couvert avec réévaluation, cette possibilité a été mise en oeuvre pour trois spécialités dont l'une a vu son ASMR réévaluée de IV à III au vu du gain démontré pour la survie des patients.
Des marges de progression sont en outre identifiées par la HAS qui déplore une sous-mobilisation de deux dispositifs :
- le pré-dépôt , qui n'a concerné en 2020 que 14 % des dossiers de primo-inscription et d'extension d'indication en procédure d'instruction complète, alors que ce processus a montré une réduction du délai à une moyenne de 78 jours ;
- la présomption d'innovation . Ce processus se distingue d'une évaluation, il s'agit d'une reconnaissance permettant d'obtenir une instruction anticipée dès la demande d'AMM. En 2020, seulement sept industriels l'ont sollicité pour dix médicaments.
Proposition n° 20 : améliorer le traitement des dossiers d'évaluation en vue de la prise en charge afin de garantir un accès rapide aux innovations.
b) Un cadre de l'accès précoce rénové
À la suite du CSIS de 2018, différentes adaptations réglementaires ont été apportées aux dispositifs français d'accès précoce, particulièrement lors de la LFSS pour 2019 avec la possibilité de délivrer des autorisations temporaires d'utilisation aux extensions d'indication et lors de la loi de financement de la sécurité sociale (LFSS) pour 2021, avec deux dispositifs : l'accès précoce et l'accès compassionnel . Cette refonte vise à clarifier les circuits d'accès dérogatoires en les réorganisant non plus d'après leur seule dénomination, mais d'après leur finalité. Il réunit l'ATU et la recommandation temporaire d'utilisation (RTU) sous trois régimes dérogatoires : accès précoce, accès compassionnel et prescription « hors AMM ».
L'ANSM considère que le dispositif adopté fin 2018 a permis un accès précoce dans des situations d'absence d'alternative satisfaisante en oncologie. Ainsi, dix-sept ATU de cohorte (ATUc) d'extension ont été autorisées en 2020, en plus des 20 ATUc octroyées avant l'AMM.
Une réforme de l'accès précoce en cours de mise en oeuvre
La loi de financement pour 2021 54 ( * ) a redéfini les dispositifs d'accès précoce, supprimant les ATU et RTU au profit de deux dispositifs nouveaux : l'accès précoce pour les médicaments innovants en développement et l'accès compassionnel quand le développement n'est pas envisagé.
Ces modifications poursuivent trois objectifs :
- répondre aux besoins d'accès aux médicaments couverts par les dispositifs actuels ;
- être plus homogène, simple et lisible pour les acteurs ;
- offrir des garanties par rapport à la soutenabilité de notre système de santé.
Cette réforme doit entrer en vigueur au 1 er juillet 2021. Elle modifie profondément la répartition des rôles entre autorités sanitaires en donnant à la HAS un pouvoir décisionnel dont elle était jusqu'ici privée. La HAS est, avec la réforme, chargée de l'autorisation des accès précoces, sur avis conforme de l'ANSM.
L'accès précoce issu de la réforme prévoit également un suivi en vie réelle avec un recueil prévu de données sur l'efficacité, les effets indésirables et les conditions réelles d'utilisation.
• L'ANSM estime que cette réforme va permettre de fluidifier les processus d'évaluation menée dans les différentes institutions (ANSM, HAS, direction de la sécurité sociale) en les rassemblant dans la même unité de temps en vue d'atteindre un accès plus rapide aux traitements et de simplifier les démarches des laboratoires, via notamment le dossier commun ANSM/HAS, sur un portail commun . Cependant, elle admet que l'efficacité du nouveau dispositif dépendra de la compréhension et de la lisibilité qu'en auront les laboratoires .
• Ce dispositif nouveau est également salué par les entreprises du médicament, qui estiment le nouveau système de financement et de prise en charge comme un modèle correspondant à leurs attentes et porteur d'une plus grande lisibilité pour les acteurs.
L'ANSM a veillé au cours des dernières années à rendre plus lisible et accessible l'accès aux produits innovants. Elle a notamment mis en place en 2019 un référentiel des spécialités disponibles en ATU nominatives (ATUn) afin d'améliorer la connaissance des prescripteurs sur la délivrance possible des molécules. Une hausse sensible du nombre de nouvelles spécialités et du nombre d'ATUn a été constatée.
Octrois d'ATU nominative
et médicaments (ou
substances actives) mis à disposition par an
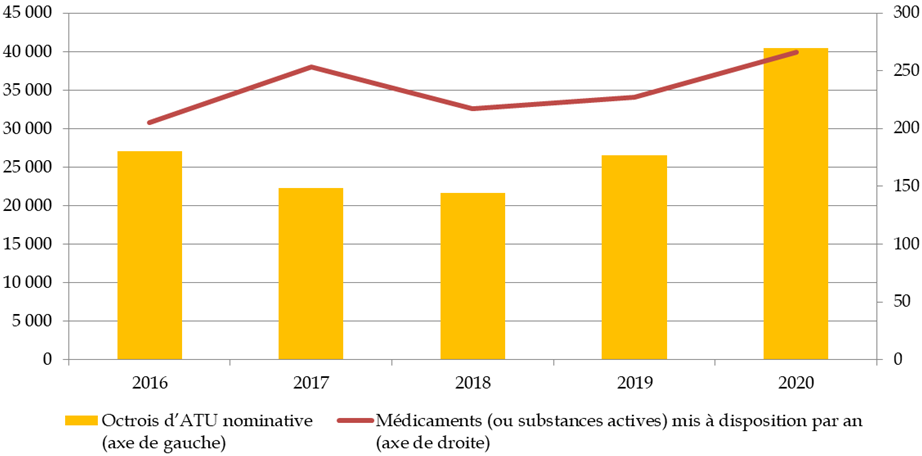
Source : Commission des affaires sociales du Sénat d'après l'ANSM
c) Des mesures attendues pour desserrer l'étau de l'accès précoce
• Des freins sont constatés en matière de médecine de précision et de médecine diagnostique , particulièrement en oncologie.
L'approche par listes montre ainsi ses limites , visibles pour ce qui est du référentiel des actes innovants hors nomenclature (RIHN). Ces enveloppes financièrement contraintes ont des conséquences néfastes sur la prise en charge des patients, en particulier ceux atteints de cancer en les privant des tests dits « compagnons » essentiels pour préciser le diagnostic et accompagner le développement de la médecine personnalisée en oncologie.
Des propositions sont faites sur ce sujet concernant les tests génétiques en oncologie , afin notamment :
- de revoir l'enveloppe du RIHN et de la liste complémentaire en ne conservant que les tests ayant réellement vocation à y figurer, en envisageant un nouveau financement pour les tests compagnons et en ayant un suivi plus fin des prescriptions et indications ;
- de redéfinir les cotations , avec notamment une dissociation d'éléments forfaitaires, liés par exemple à l'indication et au séquençage, d'une partie fonction de la complexité de l'analyse.
Proposition n° 21 : débloquer les restrictions à la prise en charge de tests génétiques en oncologie.
• Une nouvelle approche doit également être retenue à l'égard des ASMR IV.
Les améliorations ainsi classées, jugées mineures, doivent cependant bien être prises en compte. Aussi, leur prise en charge dans le milieu hospitalier n'est-elle pas assurée quand elle l'est en ville : se pose ici un problème d'accès équitable du patient à l'innovation.
S'il ne s'agit pas d'aligner la prise en charge de l'ASMR IV sur les ASMR de catégories supérieures, il est nécessaire d'intégrer les ASMR IV dans la liste en sus, ces produits représentant des innovations de rupture dont le potentiel doit être valorisé .
Proposition n° 22 : permettre l'inscription conditionnelle sur la liste en sus de thérapies innovantes avec ASMR IV, correspondant aux innovations de rupture.
En outre, interrogée sur les freins constatés dans l'accès précoce aux thérapies innovantes, l'ANSM a regretté l'absence de cadre législatif permettant la prise en charge des médicaments de thérapie innovante préparés ponctuellement (MTI PP). Une telle prise en charge serait bénéfique pour l'accès des patients à de nouvelles thérapies.
Enfin, d'autres rigidités signalées par les biotechs entendues semblent devoir être levées. Il convient de lever la condition d'inclusion de patients français dans les études cliniques à l'appui des dossiers de demande de prise en charge auprès de la HAS. Concernant les dispositifs médicaux, pour lesquels les exigences ont été relevées, il semble aussi nécessaire de disposer de davantage d'organismes en capacité de réaliser les marquages .
Proposition n° 23 : lever la nécessité de comprendre des patients français pour une étude soumise à la HAS en vue d'une prise en charge.
Proposition n° 24 : engager un plan d'action pour favoriser le développement en France d'organismes notificateurs.
2. Faire de la France un moteur de la valorisation des données de santé au service de l'innovation
a) Un potentiel à valoriser : le Health Data Hub
La création du Health Data Hub figurait parmi les mesures phares du CSIS 2018.
Si le système national des données de santé et la plateforme des données de santé ont été salués par les acteurs entendus, ceux-ci ont également rappelé le défi qui se posait dans les débats sur la réglementation et la protection des données de santé.
Alors que le secteur des données de santé devient stratégique, les acteurs regrettent la place prépondérante des grandes entreprises du numérique, souvent désignées « GAFA », avec leurs données localisées aux États-Unis. Beaucoup considèrent nécessaire que deux ou trois acteurs européens soient en capacité de gérer ce secteur.
La préoccupation relative à l'hébergement de la plateforme des données de santé par une société soumise au droit des États-Unis a été soulevée par la CNIL en 2020 55 ( * ) , soulignant la jurisprudence de la Cour de justice de l'Union européenne 56 ( * ) en matière de transfert de données. La CNIL appelait ainsi à un hébergement et une gestion uniquement soumis au droit de l'Union européenne. Le Conseil d'État a confirmé les craintes formulées par la CNIL 57 ( * ) , reconnaissant un risque de transfert de données vers les États-Unis et exigeant des garanties supplémentaires.
Les conditions de stockage, de fiabilisation et d'exploitation sécurisée sont primordiales pour assurer la confiance des patients dans l'utilisation de leurs données de santé.
Proposition n° 25 : privilégier l'hébergement des données de santé par un opérateur national ou européen.
Cependant, le principal grief fait au Health Data Hub est, pour certains, son accessibilité trop restreinte. Ainsi, la plateforme est perçue comme un outil prometteur mais dépourvu des moyens de ses ambitions. Surtout, certains acteurs regrettent que l'exploitation des données soit limitée aux projets de recherche.
Sur ce sujet, les rapporteures soulignent que, dans le contexte du transfert au système national des données de santé (SNDS) des données des systèmes d'information « SI-Dep » et « Contact » créés durant la pandémie de covid-19, la présidente de la commission, Mme Catherine Deroche, a annoncé une mission sur les données de santé au cours de la session prochaine. La présidente comme les rapporteures insistent sur la nécessité de trouver un juste équilibre entre protection des données et exploitation fructueuse de celles-ci, avec in fine des bénéfices incontestables pour les patients.
Des faiblesses ont enfin été signalées dans la constitution des données de santé et leur mise à disposition en vue d'une exploitation efficace. Ainsi, alors que la question de l'interopérabilité des données était dès 2018 désignée comme une priorité, le constat demeure d'une vraie lacune en la matière. Il est urgent d'assurer cette interopérabilité, particulièrement au niveau des établissements de santé notamment du service public hospitalier .
Proposition n° 26 : assurer l'interopérabilité des données de santé collectées par les établissements de santé pour faciliter leur exploitation dans le Health Data Hub .
b) Le suivi en vie réelle : une opportunité pour l'innovation
Le suivi en vie réelle est depuis plusieurs années montré comme une perspective d'avenir pour l'innovation en santé, à travers une capacité de développement de la médecine personnalisée, centrée sur le patient 58 ( * ) .
Les données de santé doivent être vues comme u ne chance pour la recherche médicale et donc avant tout une chance pour le patient . Un exemple marquant a retenu l'attention dans le cadre de la crise sanitaire avec la cession en Israël des données en vie réelle des personnes vaccinées contre la covid-19, celle-ci ayant été une part du contrat entre le gouvernement et le laboratoire. Celles-ci sont perçues aujourd'hui comme une opportunité pour renforcer le suivi de l'efficacité du vaccin dans la population.
La HAS estime que les données collectées en conditions réelles d'utilisation sont un enjeu majeur dans l'évaluation des produits de santé.
« En améliorant leur recueil, stockage, analyse et transparence, et plus globalement la confiance que l'on peut porter à leur résultat, la Haute Autorité de santé a la conviction que la pertinence de l'évaluation des produits de santé au service des patients sera renforcée. »
Cependant, comme l'a souligné le ministère de la recherche, les données de vie réelle ne sont aujourd'hui pas d'une qualité suffisante . Il semble primordial de conduire des évaluations basées sur des cohortes de suivi de patients traités , promues par un groupe académique afin de générer des données de qualité. Ce suivi, particulièrement déterminant dans le cas des prises en charge conditionnelle, devrait, selon le ministère, être financé par les industriels eux-mêmes.
Alors que les missions du Health Data Hub s'étendent et que des données évaluées par les patients pourraient aussi être collectées, la HAS élabore actuellement un guide méthodologique pour les études en vie réelle ; la HAS envisage également la mise en place en son sein d'un pôle spécialisé sur les données en vie réelle.
Proposition n° 27 : renforcer le suivi en vie réelle pour l'évaluation des médicaments faisant l'objet d'une prise en charge conditionnelle.
Le CSIS 2021 est une opportunité à ne pas manquer pour reconnaître enfin la santé comme un secteur stratégique pour la nation, tant pour l'efficience du système de santé qu'en termes économiques ou de souveraineté. La stratégie qui sera retenue devra avant tout être cohérente et globale, car il n'est plus envisageable de perdurer dans une politique des petits pas : l'innovation doit être appréhendée comme une chaîne de valeur continue qui part de la recherche, fondamentale puis clinique, passe par une phase industrielle compétitive, et se conclut par un accès sécurisé et un usage efficient des produits. Elle doit donc être accompagnée de façon déterminée par les pouvoirs publics sur l'ensemble de cette chaîne.
TRAVAUX DE LA COMMISSION
___________
I. Audition de MM. Marc Frouin,
directeur général de Bioserenity,
Franck Grimaud, directeur
général de Valneva,
Franck Mouthon, président de France
Biotech
et Stéphane Piat, directeur général de
Carmat
Mme Catherine Deroche . - Mes chers collègues, dans la perspective du prochain Conseil stratégique des industries de santé, le CSIS, notre commission a souhaité lancer des travaux sur l'innovation en santé dont les rapporteurs sont Annie Delmont-Koropoulis et Véronique Guillotin. Nous recueillons ce matin le témoignage d'entreprises qui se sont affrontées au processus d'accès au marché des produits innovants dans notre pays.
Nous entendons M. Marc Frouin, directeur général de Bioserenity, M. Franck Grimaud, directeur général de Valneva, M. Franck Mouthon, président de France Biotech, et M. Stéphane Piat, directeur général de Carmat
J'indique que cette audition fait l'objet d'une captation vidéo retransmise en direct sur le site du Sénat et disponible en vidéo à la demande.
Je vais vous laisser la parole pour un propos liminaire de quelques minutes chacun, avant de la passer aux rapporteurs, puis aux collègues qui souhaitent vous interroger.
Mme Delmont-Koropoulis, qui ne peut être présente ce matin, m'a fait part de ses questions, dont je vais vous donner lecture.
M. Franck Mouthon, président de France Biotech . - Je vous remercie de nous donner l'opportunité d'exprimer aujourd'hui un certain nombre de forces et de faiblesses de l'innovation en santé.
Je suis PDG co-fondateur de l'entreprise Theranexus qui est un essaimage du commissariat à l'énergie atomique. Comme souvent dans le domaine de la health tech , ce sont des solutions issues de la sphère académique qui sont passées dans le privé à travers la création de start-ups . Je suis le pur produit d'un tel transfert ; j'étais moi-même chercheur au CEA. Avant d'être président de France Biotech, je présidais un groupe de travail sur les partenariats public-privé dans le domaine de la santé, devenu observatoire du transfert de technologie.
France Biotech a vingt ans d'existence et anime le secteur de la health tech . Comme le nom ne le reflète pas nécessairement, nous avons au sein de nos adhérents des medtech, des biotechs et des solutions de la e-santé. Il y a environ 2 000 entreprises de ce secteur en France, avec à peu près 800 biotechs , un peu plus d'un millier de sociétés développant des dispositifs médicaux, le reste en e-santé qui, avec les téléconsultations et les solutions de santé digitale, a une forte croissance. On crée environ 60 entreprises dans ce domaine par an. Beaucoup de start-ups sont créées au travers des offices de transferts de technologies - les SATT, Inserm Transfert ou le CEA.
M. Franck Grimaud, directeur général de Valneva . - Je suis directeur général de Valneva, qui est une société spécialisée dans le vaccin, issue de la fusion d'une société que j'ai créée il y a vingt ans, Vivalis, et d'une société autrichienne, Intercell. Nous avons créé un nouveau pure player dans le domaine du vaccin. Nous commercialisons des vaccins dans le domaine du voyage en particulier. Nous développons le premier vaccin contre la maladie de Lyme, dont le développement est en cours avec Pfizer. Nous sommes les premiers à avoir un vaccin contre le chikungunya qui arrivera sur le marché, nous l'espérons, en 2023. Enfin, vous le savez sans doute, nous sommes en phase finale pour l'enregistrement d'un vaccin contre la covid-19 que l'on espère voir aboutir à l'automne.
La société compte aujourd'hui 600 personnes et est implantée sur différents sites en Europe. En plus de la France, l'Autriche pour la recherche, l'Écosse et la Suède pour la production, avec également une infrastructure commerciale aux États-Unis et au Canada. Je suis par ailleurs président du pôle de compétitivité du Grand ouest. Je vous remercie pour votre invitation.
M. Marc Frouin, directeur général de Bioserenity . - Je suis directeur général d'une start-up, BioSerenity, qui est issue de la Pitié-Salpetriètre. Elle a la particularité d'avoir des médecins ou infirmiers pour la moitié de ses effectifs de 650 personnes et l'autre moitié qui est technologique, centrée sur le développement des devices et l'intégration de ceux-ci dans les parcours de soins, au service des hôpitaux ou à distance. Nous sommes présents à Paris et avons une assez forte présence dans le Grand-Est. Nous disposons d'un plateau de production et de plateaux techniques au niveau transnational. 60 % de notre chiffre d'affaires est réalisé aux États-Unis. Je suis par ailleurs membre du conseil d'administration du pôle de compétitivité Medicen.
Mme Catherine Deroche , présidente . - Je vous remercie pour vos présentations. Nous savons la place que les biotechs prennent dans l'innovation et les difficultés qu'elles rencontrent.
Annie Delmont-Koropoulis avait plusieurs questions à vous poser, je me permets de les relayer.
Quel est votre sentiment sur la qualité de l'examen des demandes d'essais cliniques par l'ANSM et les CPP ? Certains industriels indiquent être parfois désarçonnés par les questions posées, voire par des décisions de refus qu'ils attribuent à une mauvaise connaissance par les experts de l'ANSM ou les membres des CPP des nouveaux mécanismes de l'innovation. Partagez-vous ce sentiment ?
La deuxième question concerne Valneva. Il semble que le Royaume-Uni se soit positionné très tôt comme un partenaire proactif de votre croissance sur toute la chaîne, de la recherche clinique à la production, avec un soutien financier significatif. Dans quelle mesure vos négociations avec les Britanniques contrastent-elles avec celles que vous aviez engagées avec la Commission européenne ? L'Europe s'est-elle positionnée uniquement comme acheteuse dans ces négociations ? Vous a-t-elle approché en 2020 pour vous accompagner dans vos développements ? Comment se fait-il que le Royaume-Uni ait « grillé la priorité » à l'Europe pour l'accès à un vaccin conçu par une biotech pourtant française ?
Lors des précédentes auditions réalisées dans le cadre de la mission, plusieurs intervenants ont fait état de difficultés dans l'accès aux financements au-delà d'un certain seuil. On parle notamment d'un « plafond de verre » pour des investissements de 500 millions d'euros, soit des montants qui, par exemple aux États-Unis, peuvent être obtenus très rapidement. Partagez-vous ce ressenti ? La durée nécessaire pour réunir les fonds utiles à vos investissements vous paraît-elle excessive ? Avez-vous des propositions à formuler pour permettre un meilleur accès des start-ups à des financements d'envergure ?
M. Franck Grimaud . - Sur la question de l'approche du sujet covid par l'Europe, en comparaison des États-Unis ou de l'Angleterre par exemple, une très bonne chose est à souligner : l'Europe a choisi de centraliser les commandes très tôt, en juillet 2020. Une autre gestion avec une compétition entre pays européens aurait été délétère. Cependant, l'Europe n'ayant pas d'outil de financement préexistant pour ce type de crises, les négociations avec les industriels se sont faites en juillet mais les premiers contrats ont été signés en septembre, octobre, novembre 2020. Or, la course pour les vaccins devait nécessairement démarrer dès le premier trimestre 2020.
Les États-Unis, qui ont un outil qu'est la Barda, avec une dotation de plusieurs milliards de dollars, ont pu financer à risque six programmes dont notamment celui de Sanofi, de Moderna, de Novavax, d'Astra Zeneca. Cela a permis de mettre sur chacun 500 millions d'euros, soit la somme nécessaire pour aller de la phase 1 à la phase 3 et financer tout ou partie de l'industrialisation. Les doses achetées au troisième et quatrième trimestres de 2020 par l'Europe avaient en réalité principalement été financées à risque par le Barda et non par l'Europe.
Nous nous sommes retrouvés dans une situation intermédiaire : du fait de notre implantation en Écosse, le Royaume-Uni a considéré que c'était un positionnement stratégique, a cru très vite en notre vaccin inactivé et a trouvé intéressant de pouvoir financer très vite. Le pays a décidé d'un financement à risque, non remboursable, de 470 millions d'euros, dont nous avons déjà reçu la moitié.
La leçon que j'en tire c'est que ce n'est pas un défaut de ressources de l'Europe : l'Europe va commander et dépenser probablement autant que les États-Unis. Le problème est un problème d'outil, il faudrait un outil équivalent au Barda.
Des discussions sont en cours pour créer une agence européenne HERA. Il faut, pour qu'elle soit effective, qu'elle soit dotée de 10 à 20 milliards d'euros. Il faudrait qu'elle soit autonome en décision, comme la Banque centrale, et n'ait pas besoin de l'avis de chacun des 27 avant de lancer des investissements et paris dans tel ou tel vaccin avec en contrepartie des doses. C'est à mon sens la leçon de l'histoire : le point positif était la centralisation des commandes, la lacune était l'absence d'agence européenne équivalente au Barda.
Nous avons aujourd'hui un financement nous permettant d'être en phase 3, nous avons un financement total pour une nouvelle usine. Nous n'en serions pas là sans soutien ; sans le Barda nous n'aurions sans doute pas été si vite. Il faut absolument une agence européenne autonome, bien dotée et en capacité de réagir très rapidement en cas de crise sanitaire. En ce qui concerne la grippe pandémique, nous avons constaté jusqu'ici des agents très infectieux mais peu pathogènes ou très pathogènes mais peu infectieux. Il y a un risque que les deux soient un jour cumulés. Une prochaine crise pandémique peut arriver ; l'Europe a tout à fait les moyens de s'y préparer, il faut les outils institutionnels pour y répondre.
M. Franck Grimaud . - En matière d'essais cliniques, nous sommes confrontés à un problème culturel. L'innovation dans le domaine des biotechnologies repose forcément sur de nouvelles approches, de nouveaux types de médicament, et emprunte souvent un nouveau mode de « delivery ». Par essence, la plupart des innovations en biotechnologie sortent des cases, de ce qui est déjà connu. Or, en France mais pas seulement, on privilégie une approche « to the book » : si on ne rentre pas dans les cases, dans des choses déjà décrites, on demande un niveau de protection maximal, alors que d'autres pays adoptent une approche plus pragmatique du rapport bénéfices-risques. Ce n'est donc pas une question d'infrastructures pour réaliser des essais, dont la France est très bien dotée. Un certain nombre de processus doivent être accélérés, mais le principal enjeu est bien culturel.
La covid-19 l'a montré : les approches ont été plus ou moins pragmatiques selon les pays et les agences, dans un contexte particulier. En termes de soutien et de rapidité dans l'accès au marché, la phase d'essais cliniques constitue un enjeu majeur pour les biotechs , car le fait de pouvoir arriver sur le marché en six ans plutôt que huit est un facteur de compétitivité déterminant. Beaucoup de sociétés françaises de biotechnologie vont réaliser leurs essais ailleurs, le problème est donc bien principalement culturel.
Les essais précliniques coûtent entre deux et cinq millions d'euros, un essai de phase 1 peut requérir quatre à cinq millions d'euros, quand un essai de phase 2 nécessite vingt millions. Pour ces étapes, on trouve des financements au travers notamment de capitaux-risqueurs. La banque publique d'investissement (BPI) est à cet égard très performante : elle a, par exemple, contribué à la fusion et la création de Valneva dont elle détient 10 % du capital.
Les biotechs françaises et européennes rencontrent cependant des blocages pour devenir les laboratoires pharmaceutiques de demain et arriver sur le marché par elles-mêmes. Nous faisons partie des rares sociétés en Europe qui disposent de leur propre réseau commercial. Le défi est de trouver le financement d'une phase 3 qui va coûter, grosso modo , entre 500 et 600 millions d'euros. Est alors requise l'intervention d'investisseurs prêts à injecter 50 à 100 millions d'euros pour une phase 3 dont le risque d'échec est de 50 %.
L'Europe compte un ou deux fonds capables de réaliser de tels investissements, contre 30 à 35 fonds aux États-Unis. C'est à ce niveau que le verrou se situe. France Biotech milite depuis dix à quinze ans pour qu'une partie de l'assurance-vie - 2 à 4 % - soit redirigée pour alimenter des fonds dotés de plusieurs milliards d'euros capables de réaliser ce type d'investissements pour accompagner les biotechs dans cette dernière phase. Il ne s'agit pas de demander des aides à l'État mais bien des incitations fiscales pour encourager cette allocation. L'enjeu est français et, plus largement, européen.
On a en Europe, à la différence des États-Unis, un problème de remboursement des médicaments. À l'heure actuelle, toutes les biotechs qui commercialisent leurs produits réalisent grosso modo 60 % de leur chiffre d'affaires aux États-Unis, avec 70 à 75 % de la profitabilité qui est réalisée dans ce pays. Cette profitabilité sert à réinvestir dans la recherche et développement (R&D). Sur dix phases 1 lancées, un seul médicament connaît le succès : le risque est donc significatif. L'Europe a, en général, opté pour une politique de remboursement économe. C'est un sujet puisque, aujourd'hui, l'essentiel de la profitabilité est réalisée aux États-Unis. Or les centres de recherche ont tendance à être localisés à proximité des clients finaux importants. C'est donc un point très sensible politiquement.
M. Stéphane Piat, directeur général de Carmat. - Carmat est une société dont on a trop et parfois mal parlé en France.
Il y a quarante ans, la covid-19 n'aurait pas frappé comme elle a frappé aujourd'hui, car on vivait jusqu'à 65 ans et les complications auraient été moindres. En traitant de mieux en mieux les gens, on crée une population fragile. Apparaissent de nouvelles pathologies, dont l'insuffisance cardiaque avancée avec la défaillance des deux ventricules, c'est-à-dire des deux chambres inférieures de notre coeur. Il s'agit d'une maladie dont on ne parlait pas il y a cinquante ans.
En repérant ce problème, le professeur Alain Carpentier, visionnaire, a révolutionné la cardiologie moderne : il s'est aperçu que les valves vieillissaient après soixante ans et que les personnes concernées mouraient. Il a alors eu l'idée d'utiliser des tissus bovins pour remplacer les valves cardiaques du corps humain. Avec cette invention, il a créé un colosse américain, le groupe Edwards, qu'une autre invention française a contribué à faire grossir.
Ce groupe américain, désormais valorisé à 80 milliards de dollars en bourse, s'est ainsi développé à partir de deux innovations françaises qui n'ont pas été soutenues. Puis le professeur Carpentier a compris que, si on répare les valves, c'est le muscle du coeur qui arrêtera de battre. Il a alors sollicité le soutien de Jean-Luc Lagardère. Trouver un franc à l'époque pour un projet très risqué porté par une start-up, c'était déjà un défi. Jean-Luc Lagardère, entrepreneur visionnaire lui-même, a accepté en lui mettant à disposition une dizaine d'ingénieurs à titre gratuit au sein de Matra. C'est l'histoire de Carmat (Carpentier-Matra).
Notre société a obtenu un agrément pour sa commercialisation, qu'on espère débuter en Allemagne. On va lancer également une étude américaine, on l'espère dans les semaines qui viennent.
Mme Catherine Deroche , présidente . - Vous êtes en négociation de prix en France ? Quelles raisons expliquent que la commercialisation démarre en Allemagne ?
M. Stéphane Piat . - La France a cette particularité de penser que le patient français est différent du patient allemand. La France réclame des études françaises à l'appui de dossiers de remboursement, alors qu'une seule étude est suffisante dans tous les autres pays d'Europe, quelle que soit l'origine du patient. Nous allons donc réaliser une étude « EFICAS » qui a été, certes, financée en partie par la Haute Autorité de santé. Dans ces conditions, le remboursement n'arrivera en France probablement qu'en 2025 ou 2026.
Un cas d'école unique dans le domaine du dispositif médical : j'ai développé un clip pour la régurgitation mitrale dans une entreprise américaine il y a dix ans. Nous avons battu le record du monde en termes de délai au remboursement. Nous avons eu deux ans de retard sur les États-Unis - pourtant déjà très lents par rapport à l'Europe pour le remboursement du dispositif médical, contrairement aux produits pharmaceutiques - qui avaient accumulé huit ans de retard sur le marquage CE. Au final, les Français ont bénéficié du clip, qui sauve la vie de milliers de gens tous les ans, avec dix ans de retard sur le marquage CE.
Le système est assez rigide pour le dispositif médical en France. Pourquoi conditionner le remboursement en France à des données sur des patients français ?
M. Franck Mouthon . - Je serai moins pessimiste sur les essais cliniques : les choses évoluent plutôt favorablement, d'abord du côté de l'agence nationale de la sécurité du médicament et des produits de santé (ANSM) pour les phases précoces. Il est en effet important que les phases précoces se réalisent en France car le choix du pays est clé pour le développement. On peut se réjouir que les choses aient progressé à l'ANSM en termes de délais et d'instruction, avec la mise à disposition d'équivalents temps plein pour l'examen des essais de phase précoce.
Le premier point d'amélioration concerne les comités de protection des personnes (CPP). Une réforme porte sur leur tirage au sort. Quand on dépose une demande d'essai clinique, l'ANSM en juge la recevabilité en examinant la sécurité du produit et le CPP examine la méthodologie et les questions éthiques. Le tirage au sort des CPP conduisait à ce qu'une demande d'essai en neurologie peut être adressée à un CPP plutôt spécialisé sur la dermatologie.
Un autre point de faiblesse est le résultat de certains scandales sanitaires : la question des conflits d'intérêts dans l'expertise. La grande crainte est que ceux qui ont travaillé avec l'industrie soient en situation de conflits d'intérêts et ne puissent participer à l'expertise. Souvent l'industrie fait appel aux gens les plus compétents, si bien qu'on peut se retrouver avec des personnes qui ne sont pas forcément les plus compétentes sur les domaines ciblés par les essais. En outre, une autre difficulté réside dans le fait que c'est au CPP de s'estimer compétent ou non. Il est toujours difficile pour un être humain de se juger non compétent dans tel ou tel domaine. On espère que les choses évolueront pour que le tirage au sort tienne compte des compétences des personnes qui sont dans ces CPP. N'oublions pas que les CPP reposent sur des bénévoles qui bien souvent sont des hospitaliers avec une charge de travail importante.
Ces éléments peuvent constituer un frein, d'abord en termes de délais dans un environnement international compétitif. Interrogés sur la réalisation d'essais de phase 2 internationaux, nos adhérents estiment que l'inclusion du premier patient français est sans doute la plus tardive par rapport à d'autres pays.
Quand on contractualise avec les centres hospitaliers, on doit établir un contrat unique valable pour tout le monde. Reste un point de faiblesse : dans l'annexe financière qui résulte d'une phase de négociation, parfois avec une dizaine de centres en France, peut prendre des délais importants. Les industriels ne sont pas fermés à la possibilité de revaloriser ce contrat unique si, en contrepartie, cela permet de réduire les délais.
Sur le financement, la mobilisation de l'épargne française est un peu l'arlésienne. Le développement d'un médicament peut nécessiter entre dix à douze ans à partir de l'identification de sa cible, et entre un et 1,5 milliard d'euros, chiffre qui tient compte de l'attrition due au taux de succès. C'est un domaine particulier pour les investisseurs. Nos sociétés sont bâties sur des jalons de création de valeur, comme la démonstration d'apport pour le patient ou encore de sécurité, et non sur des métriques financières classiques, de revenus, de chiffre d'affaires... Il faut donc acculturer les investisseurs à cette industrie.
Vous avez sans doute entendu parler du dispositif développé par Philippe Tibi, professeur d'économie de l'école poytechnique qui a écrit un essai sur le financement de la 4e révolution industrielle de la France, en passant par les technologies. En France, on a bien identifié l'enjeu des phases précoces d'amorçage qui sont désormais plutôt bien dotées. Le défi est désormais centré sur les phases de « scale up » : il manque les financements pour soutenir les étapes plus tardives de passage à la phase industrielle, notamment les phases 2b et 3 et de pré-enregistrement. Pour ces phases, des montants de plusieurs centaines de millions d'euros sont à sécuriser.
Philippe Tibi a mis en place, face à 22 assurantiels et parapublics, 33 fonds opérant dans le domaine des biotechnologies, dont neuf interviennent dans le domaine de la santé. Ces investisseurs assurantiels se sont engagés à mettre plus de six milliards dans le domaine de la technologie. Certains ont mobilisé entre 400 et 450 millions d'euros, comme le fonds Jeito, dirigée par Rafaèle Tordjman, avec une capacité d'investissement d'une cinquantaine de millions d'euros par entreprise.
Avec une telle dynamique, vous attirerez d'autres investisseurs internationaux. D'une façon générale, par rapport à d'autres domaines technologiques, les investisseurs généralistes assurantiels ont plus de mal à s'engager dans la santé car il y a besoin de cette acculturation et de mieux comprendre ce que sont les caractéristiques de notre secteur.
M. Marc Frouin . - Mon entreprise n'est pas une biotech . Elle intervient en complément des prises en charge et des traitements des patients atteints de pathologies chroniques ou dégénératives, en appui aux médecins et en sous-traitance des établissements de santé. Il s'agit de proposer aux patients des parcours de santé d'excellence.
En matière d'essais cliniques, la complexité est différente : nos essais concernent l'organisation des parcours de soins, pour accompagner les patients. Des innovations, sous la forme d'améliorations continues en petits incréments, sont également attendues sur ce secteur dans lequel les publications n'ont souvent pas de valeur en tant que telles. Nous ne sommes pas en retard en France sur ces publications, souvent suivies de guidelines déployées dans le système de soins, mais leur capture dans la médecine 2.0 ou 4.0 constitue encore un enjeu.
Il s'agit de « petits » essais, qui ne sont pas des essais lourds, mais qui sont multiples et demandent une certaine flexibilité. Il peut s'agir par exemple de valider le recours à une intelligence artificielle. Pour ces essais, nous travaillons avec les hôpitaux et les laboratoires pharmaceutiques. Avec ces derniers, les essais se déroulent principalement hors de France, essentiellement, il me semble, pour des raisons économiques alors même que les essais qui se déroulent à l'étranger peuvent être pilotés par des Français.
Sur le financement, nous rencontrons des problèmes similaires aux biotechs même si la situation est différente. Pour un entrepreneur, qui a pour objectif de créer un maximum de valeur actionnaire, c'est un privilège de gagner de l'argent. Notre modèle se rapproche plus du secteur du numérique : on peut gagner plus d'argent et pendant plus longtemps, la demande de levée de fonds est plus élevée et atteint des échelles supérieures qui se comptent, pour mon entreprise qui est dans une position intermédiaire, en centaines de millions. Il faut pouvoir travailler avec des investisseurs qui vont déclencher l'investissement des autres : c'est difficile en France même si la situation s'est améliorée. Une partie des financements peut aussi venir des clients sur certains produits qui sont immédiatement rentables. Idéalement, nous viserions un cofinancement entre capital et commande publique, qui reste difficile en France. Par exemple, tous les actes que nous pratiquons à l'hôpital sont codés, notamment dans les groupes homogènes de séjour. Toutefois, nous n'avons pas de possibilité, en tant qu'entreprise privée, de facturer directement à l'assurance maladie : il existe sur ce point un blocage de principe du ministère de la santé, qui contrôle les dépenses autant par l'offre que par les prix.
Le marché américain « paie » mieux que le marché français mais il est aussi plus concurrentiel.
M. Marc Frouin . - S'agissant de notre développement financier ( scale up ), nos besoins se chiffrent en centaines de millions d'euros. Une partie viendra des investisseurs et, à l'inverse des biotechs , une autre partie viendra de nos clients, c'est-à-dire de celles de nos activités qui sont déjà rentables. En tout cas, ce besoin reste important car la rentabilité ne se concrétise pas immédiatement dans notre secteur.
S'agissant des vaccins, idéalement, il faudrait associer un financement par le marché et la commande publique, ou a minima l'autorisation de travailler avec le secteur médical - puisqu'il faut un délai court de mise sur le marché.
Nous avons un peu de mal en France sur ce point, c'est vrai. Par exemple, nous travaillons en sous-traitance d'une centaine d'établissements de santé, tous nos actes étant codés par les établissements ou dans la facturation à l'acte. Mais afin de travailler avec toute une région, nous aurions besoin de pouvoir prendre directement le numéro de sécurité sociale des patients et de facturer l'assurance maladie, ce qui suscite un blocage de principe du ministère des solidarités et de la santé. Je comprends bien que le contrôle de nos dépenses de santé passe par le contrôle de l'offre et je ne le conteste pas ; je constate simplement que les champions mondiaux du secteur peuvent, eux, s'appuyer sur une base territoriale.
Nous faisons 60 % de notre chiffre d'affaires aux États-Unis, marché plus rémunérateur que la France mais également plus concurrentiel et plus complexe. Ainsi, nous avons passé des contrats avec quelque 500 payeurs afin de pouvoir couvrir les besoins de 250 millions d'Américains.
Enfin, s'agissant des essais cliniques, je pense qu'il faut pouvoir faire un nouveau type d'essais, en incrément, qui correspond plus au modèle du numérique. Notre système ne me semble, hélas, vraiment pas prêt à cela. Nous devons également mieux lier le financement des essais à la commande publique. En somme, avoir envie de consommer sur le territoire ce qui est produit et développé sur le territoire, comme cela a pu se faire avec les vaccins. Ce modèle devrait concerner le secteur pharmaceutique, les biotechs ou l'e-santé si l'on souhaite avoir des entreprises qui marchent.
Mme Véronique Guillotin . - Je voudrais revenir sur les essais de phases 1 et 2, que l'on dit peu développés en France. Notre pays ne figurerait qu'au cinquième rang européen, avec environ 5 % de ces essais. Pouvez-vous nous confirmer cela, par exemple dans le domaine des vaccins ou de la cardiologie et, le cas échéant, comment expliquer une telle faiblesse ? S'agit-il selon vous d'une frilosité des autorités chargées de l'examen des phases d'essai ? Constatez-vous une différence par rapport à des pays comme l'Allemagne ou la Belgique par exemple ?
En matière de valorisation de la recherche publique, avez-vous, en tant qu'industriels, été amenés à négocier avec des structures publiques de recherche pour acquérir des brevets ? Si oui, avez-vous rencontré des difficultés, notamment avec les sociétés d'accélération du transfert de technologies (SATT) ?
Enfin, au sujet de la « course au vaccin », pensez-vous que le registre national de candidats aux essais cliniques dont s'est doté le Royaume-Uni est un outil pertinent et pourrait se révéler comme un facteur d'attractivité pour un pays comme la France ?
M. Stéphane Piat . - Au sujet des essais de phases 1 et 2, je confirme que l'ANSM a changé son état d'esprit en profondeur depuis l'an dernier. Peut-être est-ce lié au changement de direction. C'est en tout cas de bon augure pour la suite.
En effet, par définition, l'innovation, c'est de l'incertitude, ce que j'avais dû expliquer à l'ancien directeur de l'ANSM dans une lettre ouverte lors de mon arrivée chez Carmat. C'est dommage, car c'est ce que l'agence doit traiter tous les jours... Dans la recherche et l'innovation, on a un dessein et un plan mais on ne sait pas ce qu'on va trouver et, bien sûr, des problèmes peuvent survenir. Face à cela, on peut soit se cloîtrer, soit réfléchir aux meilleures façons de continuer à avancer.
Donc, depuis trente ans que je travaille dans les dispositifs médicaux, je n'ai jamais fait d'étude en France car on s'y heurte à des lenteurs pour les approbations puis à des blocages incessants en cours de processus.
Je pense que cette frilosité s'explique par le manque de personnels compétents pour traiter les dossiers. Ils ne sont pas assez nombreux et ont des périmètres de compétences bien trop larges, pouvant aller, par exemple, des cosmétiques au coeur artificiel. Nos interlocuteurs n'ont donc à la fois pas assez de temps ni assez d'expertise pour être pleinement efficaces, ce qui les frustre d'ailleurs. Dès lors, soit on fait confiance et on laisse aller soit on freine, ce qui est plutôt la règle pour Carmat du fait de notre médiatisation et de l'enjeu de vie ou de mort attaché à nos produits.
De fait, lorsque nous sommes partis de France, il y a cinq ans, notre environnement a changé. Soudain, nous avions en face de nous des personnes qui savaient vraiment ce qu'est l'innovation et nous pouvions avancer sans entraves. C'est bien dommage pour nos médecins car, ainsi privés de contact avec l'innovation, ils ne peuvent participer de manière utile aux congrès et aux débats qui font avancer leur discipline.
Finalement, nous sommes revenus en France où nous avons traité deux patients. Nous avons alors eu deux petites alarmes, vraiment anodines ; cela a pourtant engendré une quantité d'appels, de réunions, etc. à n'en plus finir. Toutes choses que l'on ne retrouve pas dans les autres pays. Même si tout s'est bien fini pour nos deux patients, j'ai donc vraiment retrouvé cette frilosité qui caractérise la France, essentiellement, je le redis, parce que nous ne sommes pas assez outillés au niveau de l'expertise publique.
Au sujet des structures publiques, on veut quelque part copier les États-Unis dans le cadre du programme d'investissements d'avenir (PIA). Or les modèles sont trop différents pour que cela fonctionne bien.
Nous ne travaillons avec des laboratoires qu'en cas de nécessité. Les laboratoires publics sont à la traîne. Nous travaillons très vite et, pour eux, le rythme est différent faute de moyens.
Lorsque j'étais à San Francisco, un chercheur polytechnicien a quitté un laboratoire français : il était frustré car le maximum de financement était de 500 000 euros pour un projet de deux ans. Il est parti dans un laboratoire américain, public, au nord de San Francisco. Il avait besoin de 5 millions, on a considéré qu'il lui fallait au moins 20 millions et on lui a finalement proposé... 25 millions. C'est la différence : il y a un problème budgétaire flagrant et, en conséquence, un découragement des gens qui veulent faire de la recherche fondamentale. Les rythmes sont différent et, lorsque nous travaillons avec des laboratoires, c'est jonché de RTT...
Mme Michelle Meunier . - C'est de la caricature !
M. Stéphane Piat . - On a du mal à avoir de l'argent. On ne peut se permettre d'avoir des financements à six mois si l'on peut l'avoir à un mois ailleurs.
Avec la pandémie, tous les laboratoires étaient fermés. Nous avons failli manquer des choses très importantes et avons travaillé avec un laboratoire aux États-Unis. Il y a unproblème d'état d'esprit. On ne peut pas comparer les deux systèmes : MIT, Livermore Institute... Ce n'est pas avec des PIA que l'on va aider l'industrie à travailler avec le public. Il faut structurer le public et, quand ce sera attractif, le privé va sauter dessus !
M. Franck Mouthon . - Concernant les phases précoces (1 et 2), je reviens sur le modèle de développement des biotechs ; il n'y a pas que des enjeux de compétences. On nous demande de veiller à la sécurité des patients français et des volontaires qui entrent dans les études, les choses sont faites très sérieusement et les critères ne sont pas si différents à mon sens avec d'autres pays.
Nous avons eu avec le précédent directeur de l'ANSM la mise en place du guichet innovation, nous nous en étions félicités avec France Biotech, qui permet de bénéficier de conseils pour les entreprises. Cet aspect est fondamental pour comprendre les choix d'aller sur d'autres territoires.
Nous sommes financés, dans les premiers stades avant le premier euro de chiffre d'affaires, par des investisseurs privés. Ces investisseurs nous demandent de choisir les territoires où la valeur est la plus importante, où l'incrément de valeur est mesurable. Souvent, on nous demande de commencer nos essais cliniques aux États-Unis. La première raison est celle de la rentabilité en termes d'accès au marché de développement. Un autre aspect important est que, lorsqu'un investisseur ouvre vos données, il vous demande votre accord de licence lorsque vous développez un actif de la sphère académique et quelles ont été vos interactions avec les autorités sanitaires et en particulier l'autorité sanitaire américaine. Lorsque vous contactez la Food and Drug Administation, différentes questions sont posées et vous présentez l'indication dans laquelle vous souhaitez développer, votre produit et ce que vous en savez. Vous avez une feuille de route, partagée et engageante ; en la remplissant vous êtes sûrs de passer d'une étape à une autre. Cela diminue l'asymétrie d'information pour les investisseurs.
Cela doit être mis en relation avec ce que je disais plus tôt : il y a une dynamique entrepreunariale, 60 entreprises par an se créent. Nous avons un ministère de l'enseignement supérieur et de la recherche et un ministère des finances qui sont alignés sur cette dynamique, mais un système de santé qui n'accompagne pas ces innovations. Ce dernier les voit plutôt en fin de parcours, comme juge arbitre sur ce qu'il reste à faire et sur les seuls aspects d'autorisation de mise sur le marché et de prix.
Il y a un plan innovation et santé que nous avons fourni au CSIS avec cette préconisation : le système de santé doit revenir dans ce schéma pour accompagner l'innovation en France. Il dispose d'énormément d'informations précieuses pour conduire nos développements et les sécuriser. Il n'est pas question d'argent mais d'information. A partir du moment où vous avez la capacité de vous engager, que le régulateur, que le payeur est capable de vous accompagner du stade précoce à la mise sur le marché, cela a énormément de valeur pour les investisseurs. Cela fait partie des choses qui conduisent à aller ailleurs.
Sur la prise de risques et la maîtrise des risques : évidemment, les scandales sanitaires des dernières années en France et la chasse aux sorcières sur les conflits d'intérêts n'ont pas aidé à avoir les bonnes personnes. La question n'est pas la prise de risque mais la mesure de celui-ci. Reste enfin la question des délais, imposés par les politiques et, sur ce sujet, la France n'est pas bien placée.
Sur la valorisation de la recherche, je vous l'ai dit, je suis un pur produit du transfert de technologie, j'étais chercheur au CEA, je travaille sur le domaine des maladies à prions, maladie de Creutzfeldt-Jakob et maladie de la vache folle. Nous avons pu voir les enjeux sanitaires à ce moment et le CEA avait développé le test permettant de tester les bovins sur l'ensemble de l'Europe ; j'ai plutôt bénéficié d'un environnement favorable au transfert de technologie. Je continue d'avoir beaucoup d'entrepreneurs qui nous sollicitent, nous avons un groupe de travail sur les partenariats public-privé : nous avons invité tous les offices de transferts de technologie - SATT, Inria, il ne faut pas oublier les CHU, qui sont des acteurs incontournables de l'innovation en France
Sur la valorisation et les bonnes pratiques par rapport à d'autres écosystèmes : les SATT ont été créées pour faciliter l'ancrage local de la valorisation à plus grande proximité des chercheurs et soulager les universités ou instituts de tutelle qui n'avaient pas la capacité de valorisation. Les SATT ont permis cela, nous avons avec ces structures davantage de valorisateurs à proximité des chercheurs. En revanche, ces SATT distribuent une diversité d'innovations des radars jusqu'à un dispositif médical, en passant par un vaccin ou un test diagnostic jusqu'à une thérapie génique. C'est un grand écart en termes de compétences pour traiter les champs de valorisation. Il y a un vrai besoin : des écosystèmes sont plus ou moins performants, du fait d'un héritage de plusieurs décennies. Cela mériterait d'avoir une uniformisation vers le haut et une capacité de partage de compétences sur un sujet particulier. Dans le réseau des SATT, ce n'est pas compliqué de trouver la bonne personne pour suivre une des 60 entreprises sur un sujet précis. Nous proposons que ce réseau soit plus coordonné.
Par ailleurs, un autre sujet apparaît qui peut être une perte d'opportunité en termes de compétitivité lorsque l'on interroge les investisseurs. Lorsque vous faites appel à une SATT, vous avez la licence - actif principal de la société qui permet de définir le périmètre et les métriques financières adossées au partage de valeur en fonction de la maturité de l'actif -, la SATT a un mandat de négociation, on a une réduction de la complexité de la négociation et la SATT peut financer de la maturation de l'actif industriel et demande à l'entreprise de rembourser, ce qui peut être un frein. Les investisseurs viennent pour plusieurs années et rembourser trop tôt peu avoir un impact en termes d'attractivité. S'ajoutent à cela les prises de participation.
Licence, remboursement des frais de maturation et prises de participations peuvent conduire à un défaut d'attractivité : il faut que nous soyons dans les standards internationaux.
Mme Catherine Deroche , présidente . - Je vais passer la parole à quatre collègues qui souhaitent également vous interroger. Le temps est contraint par notre ordre du jour et l'examen d'une proposition de loi. Je vous laisserai donc répondre à l'issue des questions, et vous demanderai, en quelques mots de conclusion, vos attentes vis-à-vis du CSIS.
Mme Corinne Imbert . - Je vous remercie pour vos propos et les sujets que vous avez abordés qui nous laissent un peu pensifs...
Je vais revenir sur la question des agences. Dans la préparation du CSIS, est évoquée la création d'une agence de l'innovation en santé. M. Grimaud en a parlé et a souligné l'importance du rôle de la Barda dans la pandémie et l'intérêt de la création de l'HERA à condition qu'elle soit dotée de moyens et en capacité de réagir rapidement.
La création d'une telle agence dans notre pays vous semble-t-elle pertinente ? Quels pourraient être, selon vous, les contours de cette agence ? Quel pourrait être son positionnement pour accompagner le développement clinique et industriel des biotechs ? Comment cette agence pourrait interagir avec la future agence européenne HERA même si les sujets ne se superposeraient a priori pas ?
Pour revenir sur vos propos, j'ai noté que l'ANSM était en progrès mais qu'il y avait un manque de ressources humaines, un problème qualitatif, un manque de moyens. Est-il pertinent de créer une agence de l'innovation ou ne faudrait-il pas renforcer l'ANSM ? Je n'ai pas la réponse, votre éclairage serait utile. Si une agence de l'innovation devait être créée, de quels moyens devrait-elle disposer ?
Enfin, nous avons beaucoup insisté sur la difficulté d'avoir des procédures accélérées dans notre pays pour permettre l'accès précoce des patients à des thérapies innovantes, je n'y reviendrai pas. J'aurais une question plus spécifique sur la procédure de marquage CE est-elle aussi efficiente et protectrice sur le plan de la sécurité que les procédures d'autorisation des dispositifs médicaux aux États-Unis ? Faudrait-il faire évoluer, selon vous, la procédure spécifique aux dispositifs médicaux en Europe pour garantir un accès aussi précoce que possible aux dispositifs innovants ? On en revient sans doute au sujet de la frilosité des moyens et la question des études sur des patients français pour obtenir des remboursements.
Mme Brigitte Micouleau . - La France est engagée dans la mise en place de la plateforme des données de santé ou Health Data Hub afin d'améliorer le pilotage des ressources en données dans le champ de la santé. Ce GIP fait l'objet d'un financement public conséquent et intègre un grand nombre de data scientists. Pouvez-vous nous faire part de vos premières évaluations de cet investissement qui utilise des traitements algorithmiques d'intelligence artificielle sur des données de santé ? Quel est l'impact de la plateforme des données de santé sur vos entreprises ? Favorise-t-elle la recherche ?
Hier, sur une radio nationale, a été évoqué le sujet de la greffe du coeur. Lors de l'émission, une machine innovante a été présentée permettant la conservation d'un greffon cardiaque quelques heures supplémentaires et donc un temps de transport plus long. De quatre heures dans une glacière traditionnelle, il serait possible de conserver un coeur de six à huit heures dans une machine permettant en outre de transfuser le coeur pendant le trajet. Êtes-vous aussi enthousiaste que le journaliste à propos de l' organ care system qui se présente comme l'avenir de la transplantation cardiaque ?
Mme Annick Jacquemet . - Nous savions que la recherche avait en France des difficultés, nous en avons avec vous confirmation avec des exemples précis.
Je voudrais vous interroger sur la maladie de Lyme, vous
avez évoqué un vaccin futur. Pouvez-vous nous donner des
précisions sur les délais et les dates de mise sur le
marché ? Ma deuxième question concerne le diagnostic de la
maladie de Lyme. En médecine humaine, il y a de grandes
difficultés pour poser ce diagnostic. En tant que
vétérinaire, j'ai été surprise à plusieurs
reprises d'avoir des patients
- humains ! - qui voulaient
faire analyser leur sang dans nos laboratoires d'analyse
vétérinaire qui sont en mesure de diagnostiquer avec
précision et sûreté cette maladie. Où en est-on des
tests et du diagnostic en France ? Évoluent-ils ? Pourquoi un
tel blocage ?
Je voudrais également poser une question concernant l'attractivité de la France en matière de recherche clinique et de production biopharmaceutique qui, à mon sens doit devenir une priorité économique, en particulier avec la crise sanitaire en cours. En dehors de l'amélioration de l'attractivité des carrières des chercheurs en France pour éviter la fuite vers d'autres pays pour laquelle des financements sont à prévoir, l'environnement réglementaire doit-il également être adapté ? Quelles seraient pour vous les priorités en termes d'innovation ?
Mme Laurence Cohen . - Je vous remercie pour vos différentes interventions. Vous avez souligné que les données concernant la France dans le domaine de la recherche et du développement indiquent qu'un certain nombre d'essais cliniques ne se font pas dans l'hexagone ou, en tous les cas, ont diminué. Ne pensez-vous pas que cette baisse, outre les tracasseries administratives, est le résultat de la diminution des centres de développement ? Je pense à Sanofi, implanté dans ma ville, dont le nombre de centres est passé de 11 à 3, ce qui est problématique. Comment pensez-vous inverser les choix faits, dont les conséquences sont négatives pour tous ?
Je voulais également vous interroger sur les agences ; Corinne Imbert vous a posé des questions à ce sujet. Pour l'agence nationale de sécurité du médicament, le Gouvernement donne de plus en plus de missions sans attribuer les moyens nécessaires à leur exercice.
Je voudrais également avoir des éléments plus précis concernant votre utilisation du CIR : quel apport constitue-t-il et quelles sont les obligations et devoirs en retour ?
On constate que les essais cliniques sont de plus en plus souvent réalisés hors de France. Cette évolution n'est-elle pas le résultat de la réduction du nombre de centres de recherche et développement sur notre territoire ?
M. Franck Grimaud . - S'agissant de la maladie de Lyme, nous espérons pouvoir lancer un essai clinique de phase 3 l'année prochaine afin de pouvoir mettre un médicament sur le marché en 2025. Il y a eu de nombreuses améliorations dans la connaissance de cette maladie par les médecins mais elle demeure insuffisamment diagnostiquée.
On a tendance en France à créer trop d'organismes distincts. La BPI et l'Inserm peuvent participer à une politique favorisant l'innovation. Il serait plus pertinent de mieux doter ces acteurs existants que de créer une nouvelle agence dédiée. En ce qui concerne l'anticipation de pandémies ou de crises sanitaires, c'est au niveau de l'Europe qu'il faut agir.
Les registres nationaux de vaccination ont permis de faciliter la mise en oeuvre d'essais cliniques sur les vaccins anticovid, alors qu'ils avaient eu lieu initialement uniquement au Royaume-Uni.
Les laboratoires ont besoin de localiser leurs centres de R&D au plus près de leurs clients et des patients qui sont traités par leurs médicaments. Or, la France ne représente souvent qu'une petite partie de leur chiffre d'affaires.
Aujourd'hui la moitié des médicaments sont issus des biotechnologies. Cette proportion monte à 7 sur 10 pour les vaccins anticovid. La France est plutôt bien dotée dans ce domaine, mais les financements privés sont insuffisants.
Le Health Data Hub est une excellente idée mais il faut que les données récoltées puissent être réellement utilisées afin d'accélérer les travaux de recherche, dans le respect de l'anonymat. On a tendance à dire que les États-Unis innovent, que la Chine copie et que l'Europe règlemente. Il faut sortir de ce travers européen qui nuit à l'innovation.
M. Stéphane Piat . - La réflexion sur l'innovation ne peut faire l'impasse sur les enjeux économiques et industriels. Il faut créer un écosystème favorable.
La filière pharmaceutique pourrait disparaître en France et celle de ma medtech ne s'est jamais vraiment développée. Or, nous assistons à une concentration de ces secteurs. Dans dix ans on ne parlera plus de start-ups.
Il nous faut agir si nous souhaitons avoir une filière de santé en France.
La recherche ne doit pas avoir pour unique objectif de faire des découvertes scientifiques, c'est enjeu industriel et d'emploi. Il ne s'agit pas tant de faire de la recherche et de mettre au point des vaccins que d'avoir des résultats.
Le foisonnement des start-ups n'a pas d'intérêt si aucune ne parvient à des résultats tangibles, il faut créer des champions français. Si les financements privés manquent c'est que nous n'avons pas encore d'exemple de réussite industrielle prouvant que la France a un écosystème favorable au développement des biotechs et des medtechs .
Il convient d'arrêter de diaboliser l'industrie pharmaceutique. La clé de la réussite est dans un partenariat entre l'industrie, les médecins et l'État régulateur.
Les règles mises en place en France pour lutter contre les conflits d'intérêts, qui n'ont pas d'équivalent ailleurs, empêchent les autorités de régulation, dont l'ANSM, de faire appel aux médecins les plus compétents dans leur champ. L'ANSM ne doit pas voir honte ou peur de parler à l'industrie. Les choses évoluent d'ailleurs sur ce point. Il est préférable de renforcer les moyens des acteurs qui existent déjà plutôt que de créer une nouvelle agence.
Le CIR est une très bonne chose, il n'y aurait pas de société de medtech en France sans ce dispositif.
La machine évoquée par Mme Micouleau permet de conserver plus longtemps les coeurs prélevés sur des victimes de morts violentes, notamment dans des accidents de la route, et ainsi de maximiser les chances de transplantation réussie.
Les dispositifs de santé développés aujourd'hui sont plus complexes et perfectionnés que ceux du passé. Il faut donc des capitaux beaucoup plus importants. Un projet comme le développement d'un coeur artificiel coûte des centaines de millions d'euros.
Il y a un choix politique et stratégique à faire. Si l'on veut disposer d'une industrie en lien avec notre poids économique il faut s'en donner les moyens. En cardiologie notamment de nombreuses idées sont développées par des médecins français mais les résultats sont captés par d'autres pays qui leur offrent les conditions nécessaires.
S'agissant du marquage CE, les procédures sont solides, contrairement à ce qui a pu être dit. Le dossier Carmat équivaut à 10m 3 de papier et a donné lieu à deux ans de travail. La FDA américaine ne fait d'ailleurs pas mieux, il n'y a pas plus de rappels de produits en France qu'aux États-Unis.
Il y a une courbe d'apprentissage pour les technologies médicales comme pour les compétences d'un chirurgien. On apprend sans cesse et il faut 10 ans pour stabiliser une technologie.
Il faut accepter l'existence d'un risque et être transparent dans l'information donnée aux patients et aux professionnels de santé mais ne pas obérer toute innovation par une règlementation trop lourde.
M. Marc Frouin . - Il faudrait réfléchir un peu plus en avance de phase et définir une stratégie par rapport aux enjeux de demain.
Par exemple, on peut s'attendre au développement de dispositifs de santé de plus en plus petits, qui associent chimie et électronique et d'une médecine de précision, de plus en plus personnalisée. Il faudrait donc définir des règles adaptées au présent mais aussi à l'avenir.
Je suis d'accord pour dire qu'il faut des réussites pour attirer les investisseurs. Les premiers succès sont déterminants pour la suite. C'est parce que nous avons été parmi les premiers à faire voler des avions que notre industrie aéronautique est aujourd'hui forte.
Une de nos difficultés réside dans le fait que nous devons concilier le développement de nouvelles technologies, qui nécessitent une rémunération de l'innovation, et le financement de notre demande interne de soins, fortement socialisée.
Le Health Data Hub est une très bonne chose. Dès lors que les soins sont financés par la sécurité sociale, il est logique que les données de santé puissent être utilisées. Pour transformer l'essai il convient désormais de permettre une coopération avec l'industrie, dans le respect des données personnelles.
M. Marc Frouin . - Le Health Data Hub est un très bel outil qui s'appuie sur des équipes de premier plan. L'idée selon laquelle, à partir du moment où il s'agit de santé remboursée, les données qui en découlent doivent pouvoir entrer dans le système du Health Data Hub qui en détient la propriété, a permis de clarifier la situation. Auparavant régnait une certaine confusion sur la question de savoir si les données appartenaient au patient ou encore aux hôpitaux. Consolider des bases à partir de données couvrant la vie d'un patient est une très bonne chose.
L'anonymisation est construite et permet de conserver un tracé des patients sur la durée, quelles que soient les bases d'origine qu'on apparie, à partir d'une pseudo-virtualisation du numéro de sécurité sociale. C'est une très bonne initiative. La transformation de l'essai suppose que le système soit ouvert et puisse être mobilisé par les industriels et les porteurs de projets communs entre l'industrie et le public, et qu'on ne commence pas un projet « recherche hospitalo-universitaire » (RHU) ou un autre type de financement public sans inclusion des patients avec des retours via le Health Data Hub qui permettent de suivre leurs parcours. Le système reste embryonnaire mais est géré très intelligemment.
En revanche, un élément de faiblesse en France réside dans la standardisation du codage. Le problème ne se pose pas avec les données codées par la sécurité sociale, mais des différences notables dans la façon de saisir les données peuvent être observées dans tout le reste de la médecine, même au niveau intrahospitalier. Des efforts importants restent donc à réaliser pour répondre aux standards internationaux afin que les données qui entrent dans le système aient de la valeur. Sur cet aspect, les Américains optent pour des façons privées de faire, quand les Chinois privilégient des voies publiques. Si les données entrées dans un Health Data Hub sont toutes codées différemment, on ne saura pas les faire fonctionner.
Enfin, je perçois une grande naïveté chez le législateur comme chez le public et les médias dans l'idée que, dans le monde d'aujourd'hui, on peut anonymiser des données. Il y a tellement de richesse dans les données que, si on fait l'effort de retrouver la personne, c'est possible. Ce n'est pas un hasard si Google peut le faire. Si on croise des données, on finira par trouver des éléments assez fins pour constituer une empreinte. Le législateur pense qu'il a protégé les personnes mais, en réalité, les moyens numériques sont tels qu'on ne peut plus les protéger.
M. Franck Mouthon . - France Biotech propose la création d'une agence depuis mars 2020. Il n'est pas question d'ajouter une strate supplémentaire mais de coordonner. On s'est beaucoup inspiré de l'agence de l'innovation de défense qui a été une révolution dans ce domaine, pour organiser le passage de l'innovation jusqu'à l'achat par la direction générale de l'armement. C'est une vraie prouesse.
Une réforme du marquage CE est à l'oeuvre à partir de ce mois-ci, les critères ont été améliorés. Le problème qui va se poser est celui de l'embolisation par l'absence d'organismes notificateurs. On a rehaussé les exigences pour le dispositif médical, avec un niveau comparable aux États-Unis, mais, en face de cela, on n'a pas réuni les conditions pour que l'ensemble des entreprises puissent bénéficier de ce marquage dans un temps relativement limité. Pour le diagnostic, c'est encore pire : quand vous disposez d'un catalogue de diagnostics, vous êtes contraints de procéder produit par produit.
La donnée en santé est absolument indispensable pour l'avenir de nos entreprises, pour définir les parcours de santé. Le problème de la valorisation de la data est le suivant : on a centralisé la collecte, mais il faut que les acteurs jouent le jeu de cette collecte. Se pose la question de la rétribution de ceux qui ont collecté. Tant que le Health Data Hub n'aura pas mis en place un vecteur qui permet d'inciter ceux qui collectent sur le terrain, notamment les médecins dans différents centres hospitaliers universitaires (CHU), on continuera à rencontrer des difficultés pour centraliser et redistribuer. Il faut réfléchir à des modèles de coopération qui permettent à la fois d'entretenir cet entrepôt de données et de soutenir ceux qui collectent les données sur le terrain. On propose par exemple que soit institué un forfait d'accès à la data pour entretenir la gestion même de cet entrepôt et que se mette en place, à chaque fois qu'on demande des données, une collaboration scientifique et technique avec ceux qui les ont produites, ce qui facilitera les partenariats public-privé.
Il n'existe pas de feuille de route pour une vision stratégique offensive de l'innovation en santé en France. Ce n'est pas prévu par la loi de programmation de la recherche, et il n'y a pas de loi de programmation de la santé. On peut préférer un autre terme qu'agence, en tout cas il nous manque un chef d'orchestre, un guichet pour résoudre les problèmes. Dispose-t-on d'une vision des investissements à réaliser et dans quels domaines ?
Aujourd'hui, la psychiatrie est le premier poste de dépenses de santé. C'est pourtant le domaine dans lequel il y a le moins d'innovation en France aujourd'hui. Sinon une agence, il nous faut donc un pilote pour coordonner cette politique d'amont, qui est très bien abordée par la BPI, et cette politique d'aval où on ne parle pas ou très peu et pour laquelle les acteurs ont besoin d'avis engageants, de bénéficier d'éclairages sur la pertinence de leurs développements - question à laquelle le système de santé peut répondre, que ce soit la direction de la sécurité sociale, l'ANSM, la HAS, le comité économique des produits de santé (CEPS)... C'est pourquoi je prône la création d'une agence de l'innovation en santé à l'image de l'agence de l'innovation de défense qui ne sera rien de plus qu'un coordinateur pour permettre de régler des problèmes sur un engagement de trois mois, sur un transfert de technologie, sur la fixation du prix au niveau du CEPS, sur l'évaluation au niveau de la HAS... Il nous faut une autorité qui définisse les priorités, pour éviter le saupoudrage, et réponde aux besoins de notre système de santé dont on veut tous qu'il reste mutualiste et solidaire.
Ce point de l'ordre du jour a fait l'objet d'une captation vidéo qui est disponible en ligne sur le site du Sénat .
II. EXAMEN DU RAPPORT
Réunie le mercredi 23 juin 2021, sous la présidence de Mme Catherine Deroche, présidente, la commission examine le rapport d'information de Mmes Annie Delmont-Koropoulis et Véronique Guillotin sur l'innovation en santé.
Mme Catherine Deroche , présidente . - Nous examinons à présent le rapport d'information de Mmes Annie Delmont-Koropoulis et Véronique Guillotin sur l'innovation en santé.
Mme Véronique Guillotin , rapporteure . - Notre mission d'information sur l'innovation en santé s'est donné pour objectifs de dresser le bilan de la mise en oeuvre des mesures du dernier conseil stratégique des industries de santé (CSIS), et de préparer le prochain dont les orientations sont annoncées pour la fin du mois de juin 2021, en identifiant les principaux freins règlementaires, financiers, organisationnels mais aussi culturels qui demeurent pour stimuler l'innovation en santé en France et faciliter l'accès aux thérapies innovantes.
Si environ 80 % des mesures du CSIS de juillet 2018 ont été mises en oeuvre, certaines réformes clés restent à quai, dont celle de l'évaluation du médicament et celle de l'expertise.
Par ailleurs, la crise sanitaire a constitué un moment douloureux pendant lequel notre pays a dû prendre la mesure de ses faiblesses dans le domaine de la santé. La blessure d'égo française dans la course aux vaccins contre la covid-19, avec le triomphe de deux biotechs américaine, Moderna, et allemande, BioNTech, qui tranche avec l'échec initial de Sanofi, n'est finalement qu'un des symptômes de ce qui s'apparente de plus en plus à un déclassement de notre pays dans le développement et la production de thérapies innovantes.
Le constat partagé par l'ensemble des personnes que nous avons auditionnées est sans appel. La France a très largement sous-estimé des tendances lourdes, dont l'effacement progressif des « frontières » entre recherche fondamentale et recherche clinique et appliquée avec l'essor de la recherche translationnelle, le besoin d'investissements massifs pour prendre le virage des biotechnologies, ou encore la délocalisation des capacités de production en principes actifs. En résulte une indépendance sanitaire française sérieusement entamée.
C'est vous dire si la marche est haute quand on sait que le Gouvernement a fixé pour objectif au prochain CSIS de faire de la France la première nation européenne innovante et souveraine en santé.
Ce dernier CSIS de la mandature en cours doit donc être l'occasion pour notre pays de s'imposer, dans un environnement européen et international chaque fois plus concurrentiel, comme un moteur de l'innovation en santé dès ses phases les plus précoces, en prenant résolument le virage de la médecine personnalisée.
Mme Annie Delmont-Koropoulis , rapporteure . - Je commencerai par un axe qui nous semble déterminant si la France veut atteindre l'objectif fixé par le Gouvernement, celui de la stratégie industrielle.
Il nous semble impératif de prendre un virage clair vers les biotechnologies. Cet objectif figurait déjà dans les conclusions du CSIS de 2018, il s'agit sans plus attendre d'en faire une réalité.
Cela passe par en premier la consolidation de l'appareil industriel et donc, disons-le de manière claire, par un soutien à l'industrie pharmaceutique.
L'enveloppe contrainte de l'objectif national de dépenses de l'assurance maladie (Ondam) fait peser sur le secteur du médicament une pression particulière sur les ressources du secteur au nom de la maîtrise globale de la dépense de santé. Alors que nous entrons dans une période de forte innovation qui ouvre l'accès à des traitements coûteux, la stabilité de cette enveloppe contraint les marges des industriels sur les médicaments matures.
Il s'agit là d'un enjeu de souveraineté sanitaire. Rétablir les marges sur ces médicaments permet de préserver la production sur le territoire national ou en Europe, de maintenir des emplois voire de relocaliser. Surtout, ces chaînes de production sont autant d'atouts en temps de crise ou de tensions sur les approvisionnements.
Différentes mesures appellent à être regardées de près, au titre desquels, d'une part, un desserrement de l'enveloppe médicaments de l'Ondam, et, d'autre part, des mécanismes de prix plus sécurisants pour les investissements en faveur de la production en France de médicaments matures.
Soutenir cette ambition passe ensuite par une politique volontariste à l'égard des entreprises innovantes. Nous avons organisé au début du mois une table ronde de biotechs de laquelle nous avons retenu tant les constats forts que les recommandations pertinentes avancées.
La France est bonne pour créer des start-ups , et nous pouvons nous en réjouir. Il ne faut pas briser les dispositifs efficaces d'amorçage. Cependant, le nombre ne fait pas tout et force est de constater que les acteurs reconnaissent unanimement un « plafond de verre » dans la maturation des biotechs .
Le seul horizon d'une biotech ne peut pas être de se faire racheter par un géant américain quand ses innovations auront vocation à être mises sur le marché !
Aussi, nous considérons qu'il ne faut pas chambouler les fondamentaux du soutien à l'innovation, au premier rang desquels l'action de la Banque publique d'investissement (BPI) et le crédit d'impôt recherche (CIR). Nos recommandations vont dans une direction : dynamiser la culture du risque. Il faut accroître le capital-risque dans les investissements de la BPI mais aussi permettre, par une redirection des placements d'épargne, la constitution de fonds d'investissements capables de soutenir des projets dépassant 500 millions d'euros et même 1 milliard d'euros.
Enfin, l'environnement industriel que nous appelons de nos voeux doit être compétitif et efficace. Trop d'acteurs ont déploré un univers trop éclaté, séparant la recherche fondamentale de la recherche clinique et de la production. Trop d'acteurs ont aussi regretté un saupoudrage des financements qui ne permet pas, en définitive, de faire émerger les champions de demain. La question n'est pas l'aménagement du territoire, mais le rang que nous ambitionnons de tenir. Il faut identifier des secteurs clés, peu nombreux, sur lesquels notre pays peut tirer un avantage compétitif dans l'innovation en santé ; deux semblent indiqués : l'oncologie et l'immunologie. Il faut que l'État puisse soutenir la constitution d'un véritable cluster, atteignant une taille critique permettant d'affronter la compétition internationale.
Mme Véronique Guillotin , rapporteure . - Si la France a désormais rattrapé son retard dans la création de start-ups en santé, celles-ci nécessitent néanmoins que leur soit garantie la possibilité de détenir, à terme, la propriété intellectuelle de la découverte qu'elles comptent exploiter. C'est à cette condition que les start-ups seront en capacité d'attirer les investisseurs disposant de la surface financière suffisante pour accompagner leur maturation dans le cadre de développements cliniques de phases 2b et 3.
Nous plaidons ainsi pour la transformation progressive du mandataire unique en un véritable « propriétaire unique », pour plus de lisibilité pour les industriels et les investisseurs privés, en particulier étrangers. Ce propriétaire unique disposerait d'un mandat élargi et d'une autonomie renforcée pour la négociation et la signature de tous les contrats de transfert, y compris ceux impliquant la cession d'un résultat, sous réserve du respect d'une charte de principes partagés et d'un accord sur la répartition des revenus nets, validés par les copropriétaires dès la désignation du mandataire unique.
Par ailleurs, nous devons réagir face au décrochage préoccupant du financement public de la recherche en santé. Dans un rapport conjoint, les académies nationales de médecine et de pharmacie ont fait en mars dernier le constat d'un « recul spectaculaire du soutien à la recherche en biologie-santé » en France, avec une diminution évaluée à 25 % entre 2008 et 2020. Hors crédit d'impôt recherche, les crédits publics en recherche et développement (R&D) pour la santé en France sont plus de deux fois inférieurs à ceux de l'Allemagne. Au-delà du sous-financement structurel de la recherche publique, les organismes que nous avons auditionnés nous ont également alertées sur l'émiettement des sources de financement public qui pénalise l'accompagnement de projets à fort potentiel qui nécessitent des investissements conséquents pour poursuivre leurs développements.
Dans ces conditions, nous préconisons de doubler la part des crédits de la mission interministérielle « Recherche et enseignement supérieur » dédiés à la recherche en biologie-santé, afin d'atteindre une proportion d'au moins 30 % des financements publics en faveur de la recherche consentis à la recherche biomédicale, hors CIR. Nous appelons, en outre, à l'identification de quelques secteurs à haut potentiel et stratégiques dans le domaine de la santé et la priorisation pour ces secteurs de l'accès aux financements publics apportés par la BPI et l'agence nationale de la recherche.
Cette priorisation suppose que notre pays assume clairement le choix de se spécialiser dans certains segments de la recherche biomédicale. Ces secteurs prioritaires seraient définis par la future agence de l'innovation en santé dont nous allons préciser les contours, après concertation des acteurs de la recherche académique, hospitalière et industrielle et des acteurs du système de santé.
Mme Annie Delmont-Koropoulis , rapporteure . - Dès février 2020, la Barda américaine (« Biomedical Advanced Research and Development Authority ») a choisi d'investir dans une dizaine de projets de vaccin contre la covid-19 en soutenant chacun d'eux à hauteur de plusieurs centaines de millions de dollars. Elle s'est ainsi clairement positionnée comme une partenaire des développements, prête à assumer le risque de l'innovation. Par contraste, l'Europe s'est positionnée comme acheteuse auprès des industriels du développement de vaccins, et non comme facteur d'innovation.
Tirant les enseignements de la crise sanitaire, la Commission européenne a annoncé la création d'une autorité européenne de préparation et de réaction en cas d'urgence sanitaire, l'agence « HERA », chargée d'investir de façon réactive dans le développement et le déploiement de contremesures et l'augmentation des capacités de production. Au regard des investissements massifs que nécessitent les développements biomédicaux pour répondre à une menace sanitaire, l'enjeu est bien que l'agence européenne HERA puisse disposer d'une « force de frappe » comparable à celle de la Barda, qui se chiffre en dizaines de milliards d'euros.
En complément de cette évolution, nous sommes favorables à la création d'une agence française de l'innovation en santé qui se conçoive comme un facteur de simplification et d'accélération et non comme une strate administrative supplémentaire dans un paysage institutionnel de la recherche et de l'innovation déjà peu lisible. Afin de ne pas créer un nouvel établissement public, nous proposons de constituer l'agence de l'innovation, à l'instar de l'agence de l'innovation de défense, sous la forme d'un service à compétence nationale qui serait placé directement sous l'autorité du ministre de la santé et serait doté de pouvoirs décisionnels délégués lui ménageant une autonomie d'action sous le contrôle du ministre. Pourquoi placer ce service sous l'autorité du ministre de la santé, plutôt que sous celle du ministre de l'économie ou de la recherche ? Parce que nous estimons que le ministère de la santé doit rester pivot dans l'identification des besoins de notre système de santé qui seront pris en compte dans la définition de nos priorités stratégiques en matière d'innovation en santé.
L'agence de l'innovation en santé serait alors chargée de missions très opérationnelles. Elle devra d'abord définir, à partir d'un horizon scanning des développements de thérapies innovantes les plus prometteurs, une stratégie de spécialisation sur les segments de recherche porteurs de l'innovation en santé qui devront être soutenus prioritairement pour répondre aux besoins du système de santé.
Elle devra ensuite attribuer une présomption d'innovation à des développements et thérapies s'inscrivant dans ces segments prioritaires afin de faciliter le lancement d'un essai clinique ou un accès rapide au marché auprès de l'agence nationale de sécurité du médicament et des produits de santé (ANSM), des comités de protection des personnes (CPP), de la Haute Autorité de santé (HAS) et du comité économique des produits de santé (CEPS).
Elle devra encore servir de guichet unique pour le dépôt centralisé d'un seul et même dossier de candidature aux appels d'offres en faveur de l'innovation en santé.
Enfin, elle devra simplifier et harmoniser les procédures de transfert de propriété intellectuelle par l'élaboration d'un guide de bonnes pratiques.
En outre, la publication des décisions de présomption d'innovation permettra d'encourager le soutien des développements concernés par des investissements privés, notamment par des fonds de capital-risque, qui verront dans la présomption d'innovation accordée par l'agence la garantie d'une mise en oeuvre facilitée des essais cliniques ou d'un accès accéléré au marché.
Mme Véronique Guillotin , rapporteure . - Nos auditions nous ont également conduites à formuler une série de propositions destinées à permettre à la France de retrouver son attractivité pour accueillir la recherche clinique. La mise en oeuvre en France d'essais précoces est déterminante pour inciter un industriel à poursuivre le développement clinique et la production d'une thérapie innovante sur notre territoire. Or, en dépit de progrès observés dans la période récente, la France peine encore à se positionner en première ligne pour attirer des essais de phase précoce pour lesquels elle n'occupe que le 5 e rang européen.
Les engagements du dernier CSIS sur la réduction des délais d'autorisation des essais cliniques ont été globalement respectés par l'ANSM qui s'est mise en ordre de marche pour prioriser un certain nombre de dossiers. En revanche, les progrès dans l'instruction des demandes d'essais cliniques par les CPP restent plus mesurés. On observe d'ailleurs une dégradation du délai médian global d'évaluation à 79 jours au premier trimestre 2021, alors que le délai règlementaire d'évaluation prévoit une décision du CPP en 45 jours.
Par ailleurs, nombreux sont les promoteurs qui ont regretté une attitude très théorique et insuffisamment pragmatique des instances examinant les demandes d'essais cliniques dans l'évaluation des bénéfices-risques en France. Ils perçoivent chez les experts mobilisés par l'ANSM et les CPP une insuffisante connaissance des mécanismes des innovations de rupture qui peut les conduire à faire preuve d'une aversion disproportionnée au risque. En outre, ils déplorent une application absolutiste des règles de gestion des conflits d'intérêts qui disqualifieraient nombre d'experts disposant d'une expérience au sein de l'industrie.
Dès lors, nous plaidons pour un renforcement des capacités de la commission nationale des recherches impliquant la personne humaine (CNRIPH) afin de constituer un annuaire des experts mobilisables dans différentes aires thérapeutiques à la disposition des CPP et de mener un benchmark de la gestion des liens d'intérêt dans les autres pays pour en tirer un guide des bonnes pratiques. Nous préconisons également la mise en place d'un déontologue de l'expertise au sein de la CNRIPH qui permettrait de conseiller les CPP dans leur recours aux experts, dans un souci de préservation des principes d'impartialité, de transparence, de pluralité et du contradictoire de l'expertise sanitaire.
Enfin, nous devons réagir face à l'embolisation de l'ordre du jour des CPP par les recherches non interventionnelles de catégorie 3. La proportion de ces recherches a d'ailleurs vocation à augmenter, puisque les fabricants de dispositifs médicaux sont désormais appelés à réaliser des recherches post-commercialisation. C'est pourquoi nous prônons le transfert de l'examen des RIPH de catégorie 3 à un seul comité d'éthique qui serait spécialisé sur les recherches non interventionnelles, ainsi que le préconise la proposition de loi déposée en 2019 par notre présidente, Catherine Deroche, notre ancien collègue Yves Daudigny et moi-même.
Mme Annie Delmont-Koropoulis , rapporteure . - Le dernier axe de nos travaux fait écho à des préoccupations que nous connaissons au sein de la commission avec le développement de la médecine personnalisée. L'innovation ne peut se concevoir qu'en replaçant le patient au coeur du dispositif.
Sur ce sujet, nous considérons qu'il faut avancer sur la question des délais d'accès. Il n'est pas admissible que la France soit si mal classée sur ce point : les patients doivent avoir accès rapidement aux innovations. La question des délais porte notamment sur le temps entre l'autorisation de mise sur le marché et la mise à disposition par prise en charge. Celui-ci doit être raccourci et le délai de 180 jours enfin tenu. Cette mesure était déjà aux conclusions du CSIS de 2018, les avancées ne sont pas jugées suffisantes.
La HAS a mis en place différentes actions sur ce sujet, les exigences d'évaluation rapide doivent être renforcées.
Concernant l'accès précoce, les réformes que nous avons votées en loi de financement de la sécurité sociale (LFSS) pour 2019 et surtout en LFSS pour 2021 sont fondamentales. La réforme prévoyant les dispositifs d'accès précoce et d'accès compassionnel doit entrer en application en juillet et semble retenir l'approbation de tous : gageons que les acteurs se saisissent des nouvelles possibilités et que ce nouveau cadre apporte davantage de lisibilité.
Différents progrès sont encore néanmoins attendus, qu'il est important de souligner.
Je pense en premier lieu à la question de la médecine diagnostique, en oncologie notamment. Il est urgent de revoir la prise en charge des tests génétiques en oncologie et à cette fin de redéfinir l'enveloppe et les cotations du RIHN.
Surtout, l'approche retenue pour la prise en charge des innovations ayant une amélioration du service médical rendu (ASMR) IV (ou améliorations « mineures ») doit être revue. Il n'est pas compréhensible que ces innovations qui sont des innovations de rupture ne soient pas prises en charge à l'hôpital quand elles peuvent l'être en ville. Il faut les intégrer dans la liste en sus, selon un schéma de prix différent des autres ASMR.
Enfin, l'ensemble des acteurs s'accordent sur un point fondamental pour les enjeux d'innovation : le rôle des données de santé. Si tous ont salué la création de la plateforme des données ou Health Data Hub , il apparaît urgent de « décorseter » cet outil. Certains industriels ont regretté l'usage trop restreint à la seule recherche.
Le Health Data Hub doit devenir un réel instrument puissant au service de l'innovation. Les données sont un enjeu majeur d'évaluation des médicaments et dispositifs médicaux, avec le suivi en vie réelle qu'elles permettent. Il faut néanmoins, pour que leur exploitation soit possible et, surtout, efficace, que les données soient de qualité, ce qui n'est pas encore le cas. Des efforts doivent être faits dans la mobilisation des données, leur gestion et leur interopérabilité.
Par ailleurs, concernant ce sujet sensible, il est primordial de donner confiance. Celle-ci ne peut se gagner qu'au prix d'une plus grande fiabilisation et sécurisation de l'exploitation. Nous nous réjouissons à ce titre de la mission annoncée par la présidente Catherine Deroche sur les données de santé.
Voici mes chers collègues, l'esprit général de nos recommandations en faveur de l'innovation en santé dans la perspective du prochain CSIS. Ce CSIS est une opportunité à ne pas manquer pour reconnaître la santé comme un secteur stratégique pour la nation, tant pour l'efficience du système de santé qu'en termes économiques ou de souveraineté. La stratégie qui sera retenue devra avant tout être cohérente et globale et porter sur l'ensemble de la chaîne de valeur du médicament, car nous ne pouvons plus perdurer dans une politique des petits pas.
Mme Catherine Deroche , présidente . - Remercions Annie Delmont-Koropoulis et Véronique Guillotin, qui ont auditionné pas moins de 64 personnes pour cette mission flash, embrassant aussi bien les mondes institutionnel et médical que ceux de la recherche et de l'industrie. Il faut bien sûr fixer des règles pour ne pas tout céder aux industriels du médicament, mais bâtir avec eux une relation de confiance. Avec des règles transparentes, nous pouvons aller plus vite pour combler les retards, qui sont considérables dans certains domaines.
Mme Florence Lassarade . - Je remercie les rapporteures pour leur excellente présentation. Il est vrai que dans le cas, par exemple, du cancer du sein triple négatif, nous sommes très dépendants d'innovations thérapeutiques qui ne sont pas produites en France.
Je souhaitais vous interroger sur une autre question, peut-être hors sujet, qui m'est inspirée par une audition que nous avons conduite dans le cadre de l'office parlementaire d'évaluation des choix scientifiques et technologiques (Opecst). Un gros effort de recherche, mutualisé entre de nombreux organismes et jusqu'à l'institut français de recherche pour l'exploitation de la mer (Ifremer), a permis de détecter dans les eaux usées et avec trois semaines d'avance la survenue de vagues épidémiques. C'est en outre une technique très peu chère puisqu'un prélèvement coûte 210 euros, soit 70 000 euros par semaine.
Or le ministère de la santé est en train, sous prétexte de règles européennes, de reprendre la main et donc de faire repartir de zéro les travaux naguère soutenus par le ministère de la recherche, en confiant cette mission à Santé publique France. Est-ce une concurrence entre ministères ? C'est en tout cas une forme d'incompétence, c'est choquant, et cela me rend sceptique sur la confiance que l'on peut faire, en la matière, au ministère de la santé.
Mme Laurence Cohen . - Je remercie à mon tour les deux rapporteures pour leur travail. Je regrette toutefois qu'il ne tire pas suffisamment les leçons de la pandémie, qui a notamment montré la nécessité d'un contrôle public de l'industrie pharmaceutique, qu'il s'agisse d'innovation ou non. Les vaccins rapportent des profits colossaux aux entreprises qui les ont développés - AstraZeneca, Pfizer, Moderna, Johnson & Johnson -, alors que la recherche est en partie financée par des fonds publics, sans contrôle aucun de leur utilisation. Et le vaccin n'en est pas même considéré comme le bien commun de l'humanité... J'aurais apprécié que le rapport adopte un ton un peu plus critique, moins enclin à laisser les coudées franches au secteur privé. Il y a, au sein du Sénat, en France, en Europe et dans le monde, d'autres regards, certes minoritaires, sur ces questions. Je partage certains points sur la pesanteur administrative, mais je continue à trouver ce rapport trop univoque, trop favorable aux intérêts capitalistiques, et insuffisamment instruit de ce que nous vivons.
M. René-Paul Savary . - L'audition de Mme Kate Bingham a été remarquable : l'indépendance du privé a permis de réagir particulièrement vite.
Créer une agence sous l'égide du ministère ? Au premier abord, le mot agence m'irrite un peu, et me donne envie de demander laquelle il faut simultanément supprimer... Si c'est sous l'autorité du ministère, est-ce un service du ministère ? Cela ne me gênerait pas. Si au contraire c'est un organe privé, je crains alors que nous ayons les inconvénients des deux mondes, privé et public. Ne peut-on trouver une formule un peu différente ? Il faut que les gens dont c'est le métier aient les coudées franches. Mme Bingham parlait de « partenaire » ; il faut en tout cas trouver une formule alliant public et privé, conjuguant les avantages mais non les inconvénients des deux. Il faut en outre franchir un pas supplémentaire dans le monde des données numériques, de manière encadrée bien entendu. Je crains qu'une agence de l'innovation abritée rue de Ségur ne soit pas d'une redoutable efficacité.
M. Daniel Chasseing . - Je voudrais féliciter Mmes Delmont-Koropoulis et Guillotin, car leur rapport identifie les problèmes et proposent des solutions pour réindustrialiser notre pays. Peut-être le privé engrange-t-il trop de profits, mais la France a-t-elle seulement été capable, comme le Royaume-Uni, de mobiliser l'argent nécessaire à des recherches n'ayant que 10 % à 15 % de chances d'aboutir, comme nous l'a expliqué Mme Kate Bingham - qui avait, elle, carte blanche du Premier ministre... ?
Identifier les secteurs clés, sûrement ; doubler les crédits pour l'investissement, sans doute. Pourquoi pas faire de la recherche publique, mais la recherche privée suppose en tout cas de pouvoir prendre des risques et de rétablir les marges des laboratoires français.
Mme Laurence Cohen . - Les laboratoires pharmaceutiques sont parmi les entreprises enregistrant le plus de profits ; ce n'est pas sur eux qu'il faut pleurer...
Mme Catherine Deroche , présidente . - Le terme d'agence me gêne un peu - je l'ai toujours dit, y compris lorsque nous avons auditionné les biotechs - car il évoque une grosse machine administrative. Les acteurs du secteur, eux, songent plutôt à ce qui devrait s'appeler un comité de coordination - qui se serait toutefois attiré d'autres critiques sur le nombre de comités existants...
Quoi qu'il en soit, France Biotech a défendu la création de cette agence, les autres acteurs ont paru s'en accommoder, et le Gouvernement a l'air d'y tenir. Nous verrons à l'usage ce que cela donnera.
Mme Véronique Guillotin . - Je comprends absolument les réserves de M. Savary sur le nom de cet organe. Nous restons, en France, traumatisés par les agences, qui sont de grosses machines, souvent bureaucratiques. Elles peuvent néanmoins faciliter la détermination d'une stratégie, servir de guichet unique, simplifier les procédures et permettre d'avoir les mains plus libres pour agir... Sous la condition de pouvoir exercer de telles missions, une agence constitue un progrès. Sans doute doit-on encore nous interroger sur les missions et l'agilité des agences dans notre pays.
Madame Cohen, ce rapport ne défend en aucune façon les gains réalisés par les industries ; il part au contraire des besoins des patients, comme vous l'appelez de vos voeux. Nous nous sommes ainsi demandé comment faciliter l'accès précoce aux médicaments, et comment améliorer l'innovation et la production de médicaments sur notre territoire pour retrouver une indépendance sanitaire. Cela ne va certes pas sans ambivalences : contrôler l'activité sans la ralentir, faire une confiance raisonnable tout en produisant des marges pour investir... Je crois que le ton du rapport, centré sur le bénéfice que le citoyen français peut tirer de ce système, est au contraire équilibré.
Mme Annie Delmont-Koropoulis , rapporteure . - La crise a été révélatrice de certaines faiblesses structurelles. La délocalisation des unités de production en Asie nous a exposés à des tensions qui ont conduit à des pénuries de médicaments, notamment sur les curares utilisés dans les urgences. Nous cherchons donc à favoriser le rapatriement des moyens de production en Europe. Un autre élément d'explication des pénuries, c'est qu'en tirant à la baisse le prix des médicaments matures, l'Ondam ne favorise pas le maintien sur notre territoire des moyens de production.
S'agissant de l'agence de l'innovation, elle a été bâtie sur le même modèle que l'Agence de l'innovation de défense, qui fonctionne très bien. Cette agence de l'innovation est une demande forte du réseau des biotechs car elles ont besoin d'un acteur institutionnel légitime pour leur permettre de débloquer des situations difficiles. Il faut un chef d'orchestre pour attribuer des présomptions d'innovation à certains traitements, pour mettre en oeuvre des procédures accélérées, pour mettre en place des essais au niveau de l'Agence nationale de sécurité des médicaments et des produits de santé (ANSM) ou enfin pour permettre l'accès au marché au niveau de la Haute Autorité de santé (HAS).
Concernant le numérique en santé, il y a un double enjeu. Il s'agit, d'abord, de la collecte des données à partir des systèmes d'information des hôpitaux et il existe, ensuite, une insuffisance d'interopérabilité de ces systèmes. Il nous faut donc mettre en place des outils performants et l'exploitation qui méritera d'être évaluée. On a besoin d'un équilibre et c'est ce qui a été prévu dans cette agence.
Mme Catherine Deroche , présidente . - Le terme d'agence pose problème en santé dans la mesure où il existe déjà une multitude d'agences dans ce secteur.
Concernant l'organisation, le ministère aurait la tutelle de cette agence d'innovation en santé, qui serait un service à compétence nationale (SCN).
Mme Annie Delmont-Koropoulis , rapporteure . - L' horizon scanning permettra de repérer précocement les thérapies innovantes, les mettre en avant et réduire ainsi les délais d'accès. La présomption d'innovation va dans le même sens en favorisant l'accès à l'innovation grâce à la mise en oeuvre de procédures accélérées.
Mme Corinne Imbert . - Cela ne m'enthousiasme pas. J'ai l'impression qu'on crée une usine à gaz. J'aimerais un exemple précis de la façon dont l'agence accélérera concrètement l'innovation et permettra de gagner du temps en comparaison avec le cheminement actuel d'un médicament.
Mme Annie Delmont-Koropoulis , rapporteure . - C'est ce dont on a parlé en préparant la proposition de loi sur la médecine personnalisée. On a besoin que les procédures soient lisibles et sécurisées pour les investisseurs.
Pour répondre à Florence Lassarade, si on met l'agence sous l'autorité du ministre chargé de la santé, c'est pour s'assurer que les priorités stratégiques définies répondent aux besoins de notre système de santé. Il faut bien, de toute façon, une tutelle. La tutelle du ministère de la recherche ne serait pas suffisante et, règlementairement, il ne peut y avoir de tutelle de plusieurs ministères sur un SCN. À l'étranger, il y a des exemples d'autres tutelles pour des organismes similaires. Au Royaume-Uni, c'est surtout le côté économique qui est mis en avant.
Mme Catherine Deroche , présidente . - Ce qui est crucial, c'est quelle stratégie on donne en matière de santé. Même si c'est important, il ne faut pas se focaliser uniquement sur l'économie. Dans l'agence prévue, il y a quand même un guichet unique pour l'accès aux financements publics qui doit faciliter la vie des entreprises. À entendre les entreprises, remplir les nombreux dossiers demandés s'apparente à un parcours du combattant, comme nous avons pu le constater en nous rendant au commissariat à l'énergie atomique et aux énergies renouvelables (CEA). L'idée de France Biotech en réclamant cette agence était de mettre en place un parcours plus fluide et de simplifier les procédures au bénéfice des investisseurs mais aussi des patients. Il ne faut donc pas perdre de vue quels sont les secteurs stratégiques en santé que l'on veut prioriser, au-delà des aspects économiques.
En revanche, s'il fallait bien identifier un acteur qui chapeaute le circuit, le fait d'appeler cela une agence me semble problématique. D'autres partagent cet avis ; Unicancer ou certains laboratoires trouvent que le terme agence est perturbant.
Mme Annie Delmont-Koropoulis , rapporteure . - On a dit qu'il y avait trop d'agences en France et qu'on voulait une agence qui regroupe tout. Mais dans notre cas, il faut l'entendre comme un chef d'orchestre, un guichet unique et non pas comme une agence supplémentaire.
M. René-Paul Savary . - Dans mon idée, il y a le schéma tout public, le schéma tout privé et un schéma intermédiaire d'un partenariat public-privé qui correspondrait à notre cas. Le ministère et le secteur privé se coordonneraient et annonceraient leur part respective de financement en vue d'un résultat. On définirait néanmoins les programmes de présomption d'innovation au regard d'un avis médical.
Or, dans le cas de l'agence proposée, je crains que le privé ne soit réticent à s'impliquer en raison de la tutelle du ministère de la santé. Il faudrait innover sur l'organisation. Les investisseurs et les biotechs seront très différents selon le thème. Ce ne sont pas les mêmes spécialistes en matière d'immunologie, d'oncologie, etc . Nous avons bien vu au Royaume-Uni, sur le vaccin notamment, que cela a pu fonctionner dès lors qu'on s'entoure de spécialistes reconnus dans des domaines très spécifiques. Comment pourrait-on imaginer un meilleur partenariat public-privé à guichet unique ? Si on tombe sous la coupe du ministère de la santé, on risque de voir la haute administration tout de suite reprendre la main.
Mme Annie Delmont-Koropoulis , rapporteure . - Il faut bien une tutelle sur un organisme appelé à prendre des décisions qui engagent l'État.
Mme Véronique Guillotin , rapporteure . - L'idée de René-Paul Savary correspond à une gouvernance mixte où le public et le privé pourrait s'associer. Toutefois, dans le schéma proposé, je ne suis pas sûre qu'il faille vraiment parler de tutelle et d'agence. Peut-être devrait-on exprimer ça différemment dans le rapport ? En tout cas, l'idée de fluidifier, de définir une stratégie ou d'avoir les moyens d'agir, que l'on retrouve dans le rapport, donne la description d'un système agile et me convient bien. En France, on a un peu le traumatisme de l'agence plombante. Il faut à tout prix échapper à ça. Mais je suis convaincue qu'on avancera efficacement.
Mme Annie Delmont-Koropoulis , rapporteure . - Nous ne pouvons pas laisser les entreprises et les industriels prendre seuls des décisions qui engagent l'État. Il faut une légitimité institutionnelle pour qu'une décision gouvernementale soit prise. On n'a pas le choix.
Cette agence sera aussi destinée à la maturation des start-ups . On a vu que le mécanisme d'aide à la création des start-ups et à leur arrivée sur le marché avait bien été mis en place mais la maturation de ces entreprises rencontre encore des difficultés.
Mme Véronique Guillotin , rapporteure . - L'agence du numérique en santé (ANS), qui a été récemment créée, dépendait d'un ministère et a été efficace. Je me demande s'il ne faudrait pas insister sur les compétences et les objectifs, dont la déclinaison est intéressante, plutôt que de se polariser sur le terme d'agence et sur sa connotation.
Mme Catherine Deroche , présidente . - Comme le dit Véronique Guillotin et Annie Delmont-Koropoulis, je pense qu'il faut se concentrer sur les objectifs et les missions définis pour cet organisme. Certes le terme d'agence ne nous plaît pas, mais le fait qu'il y ait un horizon scanning des développements de thérapies innovantes, des axes prioritaires stratégiques d'investissement, un guichet unique de dépôt de candidatures ou une simplification des procédures correspond bien à ce que demandaient France Biotech et d'autres organismes. En tout cas, le privé fait ses propres investissements, ses propres recherches et ses propres essais cliniques. L'idée est ainsi de lever une grande partie des freins qui existent dans la complexité du système actuel.
M. René-Paul Savary . - En tout état de cause, le schéma est à modifier quel que soit le nom. Il faut à mon sens revenir sur la tutelle du ministère de la santé.
Avec Mmes Guillotin et Lavarde, nous avons réalisé au sein de l'Opecst un rapport provocateur préconisant la mobilisation rapide des données de santé en période de crise sanitaire. Nous pourrions imaginer que, dans l'innovation aussi, les données de tous les acteurs soient collectées et mobilisées pour servir à des fins convergentes. Il faut un mécanisme adaptable, une boîte à outils sur mesure. Mais sous l'égide du ministère de la santé, je n'y crois pas !
Mme Annie Delmont-Koropoulis , rapporteure . - Mais les décisions prises en la matière engagent l'État et les autres agences qui sont déjà sous son contrôle, comme l'ANSM par exemple.
M. René-Paul Savary . - Il faut redonner de la confiance ! Je crains qu'une agence de l'innovation placée sous le contrôle de l'État ne soit immédiatement reprise en main et ne finisse par perdre en autonomie, que cela passe par son périmètre, ou son personnel. Cela a toujours fonctionné ainsi... Je crois que nous gagnerions à proposer un mécanisme totalement novateur. Sauf erreur de ma part, ce n'est pas le cas ici.
Mme Annie Delmont-Koropoulis , rapporteure . - L'agence doit être un phare pour les investisseurs étrangers. Comment être crédible, comment attirer les investisseurs si ce n'est pas l'État qui garantit le fonctionnement de l'agence et l'effectivité de ses décisions ?
M. René-Paul Savary . - L'État peut très bien fixer le cap, mais ensuite, si je souhaite que des recherches soient conduites dans tel domaine, vers quel partenaire dois-je me tourner ? La voilà, la question essentielle !
Mme Catherine Deroche , présidente . - C'est l'État qui fixera des priorités à la HAS ou à l'ANSM, à l'évidence. Qui, sinon l'État, fixe des objectifs ? Ce qui intéresse les start-ups ensuite, c'est qu'on leur facilite les choses. Cela étant acquis, les missions de l'agence me conviennent, et la renommer « alliance » ou « pôle » ne changerait pas grand-chose à mes yeux.
Mme Corinne Imbert . - Je suis d'accord sur les objectifs que nous poursuivons tous, mais sur le rôle du ministère : attention. Il y a 25 ans, le développement du générique, encouragé pour soulager les finances sociales, vu de ma petite officine de campagne, me semblait de nature à casser la recherche en France. Confier les décisions en la matière à un ministre qui, par définition, change régulièrement, pourrait casser la dynamique. Autrement dit : quelles sont les priorités sanitaires dans notre pays, et les politiques budgétaires devront-elles toujours primer les intérêts de santé ? Mettrons-nous les moyens, quoi qu'il en coûte, au soutien des innovations qui peuvent sauver la vie des patients ?
Mme Catherine Deroche , présidente . - Le rapport en parle, qui insiste par exemple sur les médicaments matures, dont il faudra aider les producteurs à rapatrier la production sur notre territoire. Sur les objectifs de l'agence, nous sommes tous d'accord. France Biotech a défendu l'idée d'une telle agence. C'est de toute façon le CSIS qui prendra les décisions. Nous aurons l'occasion de donner notre avis lorsque des propositions concrètes nous seront faites.
Mme Catherine Deroche , présidente . - Nous pourrons affiner notre réflexion ultérieurement, en fonction des avancées réalisées à l'occasion du CSIS, pour atteindre nos objectifs communs.
Je vous propose d'autoriser la publication du rapport et de l'essentiel.
Il en est ainsi décidé.
LISTE DES PERSONNES ENTENDUES
___________
TABLE RONDE SUR L'INNOVATION EN
ONCOLOGIE
(en commun avec le groupe d'études sur le
cancer du Sénat)
La ligue nationale contre le cancer
Pr Axel Kahn , président
Institut Curie
Pr Pierre Fumoleau , directeur de l'ensemble hospitalier
Pr Dominique Stoppa-Lyonnet , chef du service de génétique médicale
Unicancer
Pr Jean-Yves Blay , président
Pr Mario Campone , président délégué
Sophie Beaupère , déléguée générale
Michael Canovas , directeur de cabinet
TABLE RONDE DE PÔLES DE COMPÉTITIVITÉ EN SANTÉ
BioValley France
Marco Pintore , directeur général
Atlanpole Biotherapies
Franck Grimaud , président
Maryvonne Hiance , vice-présidente
Jean-François Balducchi , vice-président
Florence Hallouin , directrice générale
TABLE RONDE SUR L'ÉCOSYSTÈME DE LA RECHERCHE EN SANTÉ
Institut Gustave Roussy
Pr Fabrice André , directeur de la recherche
Institut Pasteur
Dr Isabelle Buckle , executive vice-president Technology transfer & industrial Partnerships
François Romaneix , directeur général adjoint administration et finances
Louis Marty , directeur de cabinet
Inserm
Dr Gilles Bloch , président-directeur général
Anne Sophie-Etzol , chargée des relations institutionnelles
Institut hospitalo-universitaire de Bordeaux (IHU-Liryc)
Pr Pierre Jaïs , directeur général
Pr Rémi Dubois , directeur de l'innovation
Julie Boussuge-Rozé , directrice exécutive
Direction de la recherche clinique et de l'innovation de l'AP-HP
Stéphanie Decoopman , directrice
Serge Bureau , responsable du pôle Promotion
TABLE RONDE SUR LES ESSAIS CLINIQUES ET L'ACCÈS PRÉCOCE
Agence nationale de sécurité du médicament et des produits de santé (ANSM)
Christelle Ratignier-Carbonneil , directrice générale
Caroline Semaille , directrice générale adjointe chargée des opérations
Élodie Chapel , directrice Europe et innovation
Carole le Saulnier , directrice des affaires juridiques et réglementaires
Conférence nationale des comités de protection des personnes (CNCP)
Virginie Rage Andrieu , présidente
Claire Bahans , vice-présidente
François Chapuis , vice-président
Fédération des comités d'éthique de la recherche de France
Jacqueline Fagard , présidente de la fédération
Pr Marie-France Mamzer , présidente du CERAPHP Centre et du CPP d'Île-de-France
France Assos Santé
Catherine Simonin , membre du bureau de France Assos Santé et vice-présidente de la Ligue contre le cancer
Yann Mazens , collaborateur de France Assos Santé, conseiller technique produits et technologies de la santé
Christophe Demonfaucon , Association française de personnes souffrant de troubles obsessionnels compulsifs (AFTOC)
TABLE RONDE D'ACTEURS INSTITUTIONNELS
Haute Autorité de santé (HAS)
Dominique Le Guludec , présidente
Lise Alter , directrice de l'évaluation médicale, économique et de santé publique
Comité économique des produits de santé (CEPS)
Philippe Bouyoux , président
Catherine Rumeau-Pichon , vice-présidente Dispositifs médicaux
Direction générale de la santé (DGS)
Maurice-Pierre Planel , directeur général adjoint
Direction générale de la recherche et de l'innovation (DGRI)
Anne Paoletti , directrice scientifique du secteur biologie-santé
Jocelyne Berille , chargée de mission SSRI secteur biologie-santé
Corinne Borel , chargée de mission auprès du chef du service de l'innovation, du transfert de technologie et de l'action régionale (SITTAR)
TABLE RONDE SUR LES STRATÉGIES DES GRANDS
GROUPES
PHARMACEUTIQUES FRANÇAIS EN MATIÈRE
D'INNOVATION
Laboratoire Roche
Jean-François Brochard , président de Roche France
Frédéric Chassagnol , directeur Accès, affaires publiques & pharmaceutiques, directeur général délégué
Laboratoire Sanofi
Clotilde Jolivet , directrice des affaires gouvernementales et publiques France
Jacques Volckmann , directeur de la recherche & développement France
Laboratoire Novartis
Frédéric Collet , président de Novartis France
Lamia Boudiaf , directrice médicale Oncologie
Les entreprises du médicament (Leem)
Philippe Lamoureux , directeur général
Thomas Borel , directeur Recherche, innovation, santé publique et engament sociétal
Laurent Gainza , directeur Affaires publiques
Syndicat de l'industrie du diagnostic in vitro (Sidiv)
Isabelle Tongio , présidente
Pascale Cousin , directrice générale
Caroline Boulvin , directrice générale adjointe
Syndicat de l'industrie chimique organique de synthèse et de la biochimie (Sicos)
Vincent Touraille , président du Sicos et directeur de la stratégie EuroAPI/Sanofi
Gildas Barreyre , administrateur du Sicos, vice-président de l'EFCG et directeur Énergie et affaires publiques Seqens
Sébastien Rose , administrateur du Sicos et vice-président R&D Axyntis
Catherine Lequime , déléguée générale du Sicos
PERSONNALITÉS QUALIFIÉES PILOTANT LES TRAVAUX DU CSIS
Agnès Audier , spécialiste des enjeux de transformation notamment digitale, en particulier concernant l'industrie 4.0 ou «industrie du futur» et de la transformation des services publics, ancienne directrice associée au Boston Consulting Group
Muriel Dahan , membre de l'Académie nationale de pharmacie, inspectrice IGAS, membre de la task force interministérielle vaccins
Lyse Santoro , directrice générale de la société THAC, membre du HCERES
* 1 Présentées par le Premier ministre le 10 juillet 2018.
* 2 Ces types de recherche sont désormais fortement interconnectés et forment un continuum jalonné d'allers-retours fortement facilités par la maximisation de l'accès aux données de santé.
* 3 Qui devrait s'incarner par le lancement du portail européen centralisé de dépôt des demandes d'essais à la fin de l'année 2021.
* 4 Règlement (UE) n° 536/2014 du Parlement européen et du Conseil du 16 avril 2014 relatif aux essais cliniques de médicaments à usage humain et abrogeant la directive 2001/20/CE.
* 5 Règlement (UE) n° 2017/745 du Parlement européen et du Conseil du 5 avril 2017 relatif aux dispositifs médicaux, modifiant la directive 2001/83/CE, le règlement (CE) n° 178/2002 et le règlement (CE) n° 1223/2009 et abrogeant les directives du Conseil 90/385/CEE et 93/42/CEE.
* 6 Arrêté du 27 avril 2020 relatif aux investissements étrangers en France.
* 7 Audition de Mme Kate Bingham par la commission des affaires sociales du Sénat, 16 juin 2021.
* 8 Décret n° 2014-1518 du 16 décembre 2014 relatif au mode de désignation et aux missions du mandataire prévu à l'article L. 533-1 du code de la recherche.
* 9 Ce mandataire est en théorie chargé de l'ensemble des activités de gestion et de valorisation des résultats d'une découverte issue de personnes publiques investies d'une mission de recherche.
* 10 Loi n° 2019-486 du 22 mai 2019 relative à la croissance et la transformation des entreprises.
* 11 Décret n° 2020-24 du 13 janvier 2020 relatif à la gestion de la copropriété des résultats de recherche, au mode de désignation et aux missions du mandataire unique prévu à l'article L. 533-1 du code de la recherche.
* 12 Le décret du 13 janvier 2020 a bien habilité le mandataire unique à procéder, à titre exclusif, à la négociation et la signature de tous les contrats de transfert y compris ceux impliquant la cession d'un résultat dans des conditions définies préalablement par les copropriétaires. Cette concertation préalable permet d'encadrer le pouvoir de cession du mandataire unique.
* 13 Voire la gestion de contrats industriels préfigurant la gestion de la propriété intellectuelle.
* 14 Réponses de l'institut Pasteur au questionnaire de la mission d'information.
* 15 Organisme non lucratif (« medical research charity »).
* 16 Des demandes de reversement à une SATT de près de 50 % du chiffre d'affaires issu de l'exploitation d'une technologie transférée peuvent logiquement déconcerter les acteurs du tissu économique local lorsqu'est en jeu une application relativement modeste au démarrage.
* 17 Données transmises par la direction générale de la recherche et de l'innovation (DGRI) du ministère de l'enseignement supérieur, de la recherche et de l'innovation (MESRI).
* 18 Les crédits publics en R&D (hors CIR) pour la santé en France sont plus de deux fois inférieurs à ceux de l'Allemagne.
* 19 Académie nationale de médecine et académie nationale de pharmacie, Réformer la recherche en sciences biologiques et en santé - Partie I, le financement , rapport bi-académique, 30 mars 2021.
* 20 Loi n° 2020-1674 du 24 décembre 2020 de programmation de la recherche pour les années 2021 à 2030 et portant diverses dispositions relatives à la recherche et à l'enseignement supérieur.
* 21 Les dotations des programmes investissements d'avenir dans le domaine de la santé s'élèvent à 2,6 milliards d'euros depuis 2011.
* 22 Dont quatre déjà identifiées par le Gouvernement et onze en cours d'élaboration et de consultation.
* 23 La santé digitale ; les biothérapies et la bioproduction de thérapies innovantes ; les maladies infectieuses émergentes - menaces nucléaires, radiologiques, biologiques et chimiques.
* 24 Selon le site de l'agence nationale de la recherche, « les PEPR exploratoires visent des secteurs en émergence avec des travaux de recherche dont les domaines d'application peuvent, pour certains, relever encore d'hypothèses de travail. Il s'agit d'explorer des champs scientifiques dont les retombées espérées peuvent être multiples. »
* 25 Qui constitue un bureau du département américain fédéral de la santé.
* 26 Audition du 16 juin 2021.
* 27 « UK Vaccin Taskforce ».
* 28 Cette initiative, qui doit se traduire prochainement par la publication par la Commission européenne d'une proposition de règlement, fait suite au souhait exprimé par sa présidente lors de son discours sur l'état de l'Union européenne de 2020 de constituer une « Barda européenne », qui a débouché sur une communication de la Commission en novembre 2020 pour la construction d'une Union européenne de la santé (communication de la Commission au Parlement européen, au Conseil, au Comité économique et social européen et au Comité des régions, « Construire une Union européenne de la santé : renforcer la résilience de l'UE face aux menaces transfrontières pour la santé », COM(2020) 724 final, 11 novembre 2020).
* 29 Dotée d'un budget d'1,2 milliard d'euros en 2019 qui devrait être porté à plus d'1,5 milliard d'euros en 2022.
* 30 Le responsable de ce service disposerait ainsi d'une délégation de pouvoir et d'une autonomie plus importante que dans l'hypothèse où le SCN aurait été rattaché à un directeur d'administration centrale du ministère de la santé.
* 31 Le secrétariat général pour l'investissement (SGPI).
* 32 Le ministère de l'enseignement supérieur, de la recherche et de l'innovation (MESRI).
* 33 Qu'il s'agisse d'essais sur des médicaments, des MTI ou d'essais hors produits de santé.
* 34 Décret n° 2021-301 du 19 mars 2021 modifiant certains articles du titre II du Livre I er de la première partie du code de la santé publique (partie règlementaire) relatif aux recherches impliquant la personne humaine.
* 35 Loi n° 2020-1576 du 14 décembre 2020 de financement de la sécurité sociale pour 2021.
* 36 http://www.leem.org/recherche-et-developpement .
* 37 Table ronde sur l'innovation en santé du 2 juin 2021
* 38 Didier Truchet, « Déontologie des experts en santé, perspectives critiques », Revue de droit sanitaire et social , Sirey, Dalloz, 2018.
* 39 Principes énumérés à l'article L. 1452-1 du code de la santé publique, dans sa rédaction résultant de la loi n° 2011-2012 du 29 décembre 2011 relative au renforcement de la sécurité sanitaire du médicament et des produits de santé.
* 40 Article 31 de la loi n° 2020-1525 du 7 décembre 2020 d'accélération et de simplification de l'action publique.
* 41 En prévoyant la remise d'un dossier comprenant des attestations de conformité à la règlementation et aux référentiels de la commission nationale de l'informatique et des libertés (CNIL) sur les traitements de données.
* 42 Proposition de loi relative à l'évaluation éthique de la recherche impliquant la personne humaine, déposée au Sénat le 7 novembre 2019.
* 43 Qui jouent un rôle déterminant dans l'animation des CPP et la fixation de leur ordre du jour.
* 44 Loi n° 2012-300 du 5 mars 2012 relative aux recherches impliquant la personne humaine.
* 45 Notamment des recherches en neurologie ou sciences cognitives, comme les recherches relatives à l'« homme augmenté ».
* 46 Plus large qu'un protocole de recherche, il est défini comme un ensemble d'activités de recherche organisées en vue de faciliter et d'accélérer les découvertes dans un domaine scientifique déterminé.
* 47 Dont la dotation est incluse dans les missions d'intérêt général d'aide à la contractualisation (Migac).
* 48 Ministère des solidarités et de la santé, « Évolution du modèle de répartition de la dotation socle au titre des Merri », avril 2021.
* 49 87 % en 2019 contre 54 % en 2018 pour l'étape « ANSM » et 33 % en 2019 contre 25 % en 2018 pour l'étape « CPP ».
* 50 Directive 2001/83/CE du Parlement européen et du Conseil du 6 novembre 2001 instituant un code communautaire relatif aux médicaments à usage humain
* 51 Rapport d'activité 2019 - septembre 2020.
* 52 Dossier du LEEM - Accès au marché, octobre 2020.
* 53 Ce délai concerne les nouveaux médicaments ayant reçu une première autorisation de mise sur le marché entre 2016 et 2019, en excluant les génériques et biosimilaires. Ce délai représente la période entre l'obtention de l'AMM et le remboursement effectif.
* 54 Article 78 de la loi n° 2020-1576 du 14 décembre 2020 de financement de la sécurité sociale pour 2021.
* 55 Commission nationale de l'informatique et des libertés
* 56 Arrêt du 16 juillet 2020, dit « Schrems II ».
* 57 Ordonnance du 13 octobre 2020.
* 58 Voir notamment le rapport de Bernard Bégaud, Dominique Polton et Franck von Lennep : Les données de vie réelle, un enjeu majeur pour la qualité des soins et la régulation du système de santé - l'exemple du médicament (2017).