







Rapport d'information n° 569 (2017-2018) de M. Yves DAUDIGNY , Mmes Catherine DEROCHE et Véronique GUILLOTIN , fait au nom de la mission d'évaluation et de contrôle de la sécurité sociale et de la commission des affaires sociales, déposé le 13 juin 2018
Disponible au format PDF (2,4 Moctets)
Synthèse du rapport (451 Koctets)
-
AVANT-PROPOS
-
SYNTHÈSE : LE PARCOURS D'ACCÈS
AUX MÉDICAMENTS
-
LISTE DES PROPOSITIONS
-
EXPOSÉ GÉNÉRAL
-
I. CONSOLIDER LES AUTORISATIONS TEMPORAIRES
D'UTILISATION (ATU), FORCE DU MODÈLE FRANÇAIS D'ACCÈS
PRÉCOCE AUX MÉDICAMENTS INNOVANTS
-
A. UN DISPOSITIF PLÉBISCITÉ PAR
L'ENSEMBLE DES ACTEURS DE LA SANTÉ, QUI A CHANGÉ DE NATURE
DEPUIIS SA CRÉATION
-
1. Des principes garantissant un accès
précoce et universel aux médicaments innovants
-
a) Un dispositif conçu comme temporaire et
dérogatoire, visant à répondre aux situations d'impasse
thérapeutique pour les patients atteints de pathologies graves ou
rares
-
b) Un dispositif permettant un accès large
à l'innovation
-
(1) Les ATU de cohorte permettent de traiter un
grand nombre de patients chaque année selon une procédure
équitable et transparente
-
(2) Les ATU nominatives permettent de contourner
les aléas de la mise à disposition des produits de santé,
liés notamment à la stratégie industrielle des
laboratoires
-
c) Un dispositif financé par l'assurance
maladie : un élément d'attractivité
indéniable
-
d) Un dispositif qui reste unique en Europe
-
a) Un dispositif conçu comme temporaire et
dérogatoire, visant à répondre aux situations d'impasse
thérapeutique pour les patients atteints de pathologies graves ou
rares
-
2. Un dispositif qui a changé de nature
depuis sa création
-
1. Des principes garantissant un accès
précoce et universel aux médicaments innovants
-
B. PRÉSERVER L'ATTRACTIVITÉ ET
L'EFFICACITÉ DU SYSTÈME DES ATU EN L'ADAPTANT AUX
RÉALITÉS NOUVELLES DE L'INNOVATION
-
1. Améliorer le suivi du dispositif pour en
accroître la réactivité
-
2. Répondre aux situations de rupture
d'équité entre patients : améliorer la
continuité des prises en charge
-
3. Les extensions d'indication, « trou
dans la raquette » du dispositif : remédier à un
besoin thérapeutique urgent
-
a) Un cadre juridique inadapté aux nouveaux
enjeux liés au mode d'action des innovations oncologiques
-
(1) Les ATU sont délivrées par
produit et non par indication
-
(2) Ce régime rencontre aujourd'hui ses
limites scientifiques, entraînant d'inacceptables pertes de chances pour
les patients
-
(3) Le dispositif des recommandations temporaires
d'utilisation (RTU) n'apporte pas une réponse adaptée à ce
besoin
-
b) Adapter les ATU pour couvrir les besoins
thérapeutiques liés aux extensions d'indication
-
a) Un cadre juridique inadapté aux nouveaux
enjeux liés au mode d'action des innovations oncologiques
-
4. Revenir sur un régime de
régulation financière devenu excessivement complexe
-
1. Améliorer le suivi du dispositif pour en
accroître la réactivité
-
A. UN DISPOSITIF PLÉBISCITÉ PAR
L'ENSEMBLE DES ACTEURS DE LA SANTÉ, QUI A CHANGÉ DE NATURE
DEPUIIS SA CRÉATION
-
II. FLUIDIFIER L'ACCÈS AUX
MÉDICAMENTS INNOVANTS APRÈS LEUR AUTORISATION DE MISE SUR LE
MARCHÉ
-
A. AU-DELÀ DES LOURDEURS
RÉGLEMENTAIRES, UN CLIMAT DE DÉFIANCE ET D'INCERTITUDES
-
1. L'accès au marché de droit commun
des médicaments : une procédure séquencée et
à forts enjeux
-
a) L'autorisation de mise sur le
marché : un examen centralisé au niveau européen pour
les médicaments innovants
-
b) Un cadre national rigoureux en vue de la prise
en charge des médicaments par la solidarité nationale
-
(1) La phase d'évaluation par la Haute
Autorité de Santé (HAS) : une procédure lourde
mais essentielle
-
(2) La phase de fixation du prix : une
démarche conventionnelle qui s'inscrit dans un cadre budgétaire
contraint
-
(3) Des choix d'organisation différents
entre pays
-
a) L'autorisation de mise sur le
marché : un examen centralisé au niveau européen pour
les médicaments innovants
-
2. La France à la traîne de
l'Europe ?
-
3. Quels sont les limites et les freins
identifiés ?
-
1. L'accès au marché de droit commun
des médicaments : une procédure séquencée et
à forts enjeux
-
B. INVENTER DES RÉPONSES NOUVELLES POUR
MIEUX ANTICIPER, ACCUEILLIR ET ACCOMPAGNER LES INNOVATIONS
-
A. AU-DELÀ DES LOURDEURS
RÉGLEMENTAIRES, UN CLIMAT DE DÉFIANCE ET D'INCERTITUDES
-
III. GARANTIR L'ÉQUITÉ
D'ACCÈS AUX TRAITEMENTS INNOVANTS
-
A. LES MÉDICAMENTS ONÉREUX À
L'HÔPITAL : RÉFORMER UN MODE DE PRISE EN CHARGE AUJOURD'HUI
FRAGILISÉ
-
B. PRENDRE EN COMPTE LES ENJEUX ÉMERGENTS
RELATIFS AU FINANCEMENT DES ACTES DE BIOLOGIE INNOVANTS
-
A. LES MÉDICAMENTS ONÉREUX À
L'HÔPITAL : RÉFORMER UN MODE DE PRISE EN CHARGE AUJOURD'HUI
FRAGILISÉ
-
IV. CONFORTER LE RÔLE DES ESSAIS CLINIQUES
DANS L'ACCÈS PRÉCOCE DES PATIENTS AUX TRAITEMENTS
INNOVANTS
-
A. LA RECHERCHE CLINIQUE COMME VOIE D'ACCÈS
PRÉCOCE AUX TRAITEMENTS INNOVANTS
-
B. LA NECESSITÉ D'ACCÉLÉRER
L'ACCÈS DES PATIENTS AUX ESSAIS CLINIQUES DANS DES CONDITIONS OPTIMALES
DE SÉCURITÉ
-
C. « L'UTILISATION TESTIMONIALE
ECLAIRÉE ET SURVEILLÉE » :
UN DISPOSITIF QUI SOULÈVE DES ENJEUX D'ÉTHIQUE
ET DE SECURITÉ
-
A. LA RECHERCHE CLINIQUE COMME VOIE D'ACCÈS
PRÉCOCE AUX TRAITEMENTS INNOVANTS
-
I. CONSOLIDER LES AUTORISATIONS TEMPORAIRES
D'UTILISATION (ATU), FORCE DU MODÈLE FRANÇAIS D'ACCÈS
PRÉCOCE AUX MÉDICAMENTS INNOVANTS
-
EXAMEN EN COMMISSION
-
LISTE DES PERSONNES ENTENDUES
-
LISTE DES DÉPLACEMENTS
N° 569
SÉNAT
SESSION ORDINAIRE DE 2017-2018
|
Enregistré à la Présidence du Sénat le 13 juin 2018 |
RAPPORT D'INFORMATION
FAIT
au nom de la mission d'évaluation et de contrôle de la sécurité sociale (1) de la commission des affaires sociales (2) sur l' accès précoce à l' innovation en matière de produits de santé ,
Par M. Yves DAUDIGNY, Mmes Catherine DEROCHE et Véronique GUILLOTIN,
Sénateurs
|
(1) Cette mission d'évaluation est composée de : M. Jean-Noël Cardoux, Président ; Mme Michelle Meunier, M. Jean-Marie Vanlerenberghe, Vice - Présidents ; MM. Michel AMIEL, Daniel Chasseing, Mme Véronique Guillotin, M. Dominique Watrin, Secrétaires ; MM. Bernard Bonne, Yves Daudigny, Gérard Dériot, Mmes Catherine Deroche, Élisabeth Doineau, Corinne Féret, Pascale Gruny, MM. Alain Milon, René-Paul Savary. (2) Cette commission est composée de : M. Alain Milon , président ; M. Jean-Marie Vanlerenberghe , rapporteur général ; MM. René-Paul Savary, Gérard Dériot, Mme Colette Giudicelli, M. Yves Daudigny, Mmes Michelle Meunier, Élisabeth Doineau, MM. Michel Amiel, Guillaume Arnell, Mme Laurence Cohen, M. Daniel Chasseing , vice-présidents ; M. Michel Forissier, Mmes Pascale Gruny, Corinne Imbert, Corinne Féret, M. Olivier Henno , secrétaires ; M. Stéphane Artano, Mmes Martine Berthet, Christine Bonfanti-Dossat, MM. Bernard Bonne, Jean-Noël Cardoux, Mmes Annie Delmont-Koropoulis, Catherine Deroche, Chantal Deseyne, Nassimah Dindar, Catherine Fournier, Frédérique Gerbaud, M. Bruno Gilles, Mmes Nadine Grelet-Certenais, Jocelyne Guidez, Véronique Guillotin, Victoire Jasmin, M. Bernard Jomier, Mme Florence Lassarade, M. Martin Lévrier, Mmes Marie-Noëlle Lienemann, Monique Lubin, Viviane Malet, Brigitte Micouleau, MM. Jean-Marie Mizzon, Jean-Marie Morisset, Philippe Mouiller, Mmes Frédérique Puissat, Laurence Rossignol, Patricia Schillinger, M. Jean Sol, Mme Claudine Thomas, M. Jean-Louis Tourenne, Mme Sabine Van Heghe, M. Dominique Watrin . |
AVANT-PROPOS
Mesdames, Messieurs,
Au début des années 1990, la France s'est dotée d'une politique ambitieuse, pionnière et volontariste en matière d'accès précoce des patients français aux médicaments innovants . Le dispositif des autorisations temporaires d'utilisation (ATU) a permis un accès large et rapide de patients atteints de maladies graves, sans alternative thérapeutique, à de nouveaux médicaments avant leur autorisation de mise sur le marché (AMM), c'est-à-dire plusieurs mois voire plusieurs années avant que ces molécules soient accessibles dans d'autres pays.
À côté de cet outil formidable, notre pays dispose de solides atouts, largement reconnus à l'international : l' excellence de sa recherche clinique et de ses équipes médicales ; l'attachement fort à un système de prise en charge solidaire des dépenses de santé ancré sur les principes d' équité et d'universalité dans l'accès aux soins , y compris les plus onéreux, d'un bout à l'autre du territoire ; le haut niveau d'exigence de nos agences sanitaires qui garantit la sécurité des soins que nos concitoyens sont en droit d'attendre.
Ce « modèle français » d'accès précoce aux innovations, qui a fait ses preuves, est-il aujourd'hui toujours aussi robuste et exemplaire ?
Des signaux d'inquiétude se font entendre. Les patients français auraient-ils désormais un accès plus lent à certains produits innovants - c'est-à-dire susceptibles de constituer un progrès thérapeutique ?
Si les discours sont plus ou moins nuancés, les travaux engagés par vos rapporteurs ont confirmé combien cette question est devenue cruciale pour l'ensemble des acteurs de la santé, non seulement les entreprises pharmaceutiques mais également les pouvoirs publics, la communauté médicale et les représentants des patients. Des réflexions sont ainsi en cours, notamment dans le cadre du huitième conseil stratégique des industries de santé (CSIS) 1 ( * ) dont les conclusions sont attendues au début du mois de juillet.
Le contexte et les enjeux ont profondément changé et viennent bousculer ou réinterroger, en France comme ailleurs, la façon dont nous accueillons l'innovation au sein du système de santé.
L'accélération des innovations est un puissant moteur de cette prise de conscience : de nouvelles thérapies, en particulier pour le traitement du cancer (immunothérapie, médecine personnalisée...) ou des maladies rares, offrent la promesse formidable d'améliorer les taux de survie de patients atteints de maladies graves ainsi que leur qualité de vie.
Dans d'autres domaines, le potentiel est inouï : comme l'a indiqué Marc Peschanski lors d'une table ronde sur les cellules souches organisée au Sénat le 16 mai 2018 par le président de la commission des affaires sociales, « nous avons changé de registre car nous pouvons maintenant envisager le passage de la recherche vers l'application à la médecine. Cette médecine régénératrice dont on parle depuis des années est à portée de main. »
On peut alors vouloir faire bénéficier les patients, le plus tôt possible, dans des situations d'urgence vitale, d'une chance supplémentaire. C'est dans cette ambition légitime que s'est inscrit le débat tenu au Sénat, lors de l'examen du PLFSS pour 2018, sur un amendement présenté à l'initiative de notre collègue René-Paul Savary visant à permettre le recours volontaire de patients incurables à de nouvelles molécules à des stades très précoces de leur développement. L'attente, les lenteurs, les freins sont en effet insupportables non seulement pour les malades et leur famille, mais également pour les équipes médicales dont la vocation est de soigner .
Dans le même temps, l'accélération de l'accès des patients aux médicaments innovants est en tension avec l'impératif de sécurité d'une part, et avec le défi de la soutenabilité financière de notre système d'assurance maladie d'autre part, en raison de la hausse inédite du prix des innovations thérapeutiques dans un cadre budgétaire contraint.
Dans cet équilibre délicat, le modèle français demeure sans doute parmi les meilleurs au monde mais il paraît aujourd'hui fragilisé.
Fragilisé dans sa capacité à répondre correctement à l'ensemble des besoins thérapeutiques urgents, avec un système à certains égards en décalage par rapport à la réalité des thérapies innovantes.
Fragilisé dans sa capacité à accompagner avec souplesse l'innovation et à garantir un accès équitable, avec un rapport de force parfois paralysant entre industriels et pouvoirs publics autour des enjeux de régulation du prix du médicament et des procédures insuffisamment fluides.
Fragilisé, encore, dans sa capacité à maintenir l'attractivité de la France sur une scène internationale de plus en plus concurrentielle.
Par leurs propositions, qui concernent l'ensemble de la chaîne d'accès des patients aux médicaments innovants - des essais cliniques à la commercialisation en passant par les ATU - vos rapporteurs souhaitent contribuer utilement au débat et fixer le cap des orientations à prendre pour un accès aux innovations pleinement efficace 2 ( * ) .
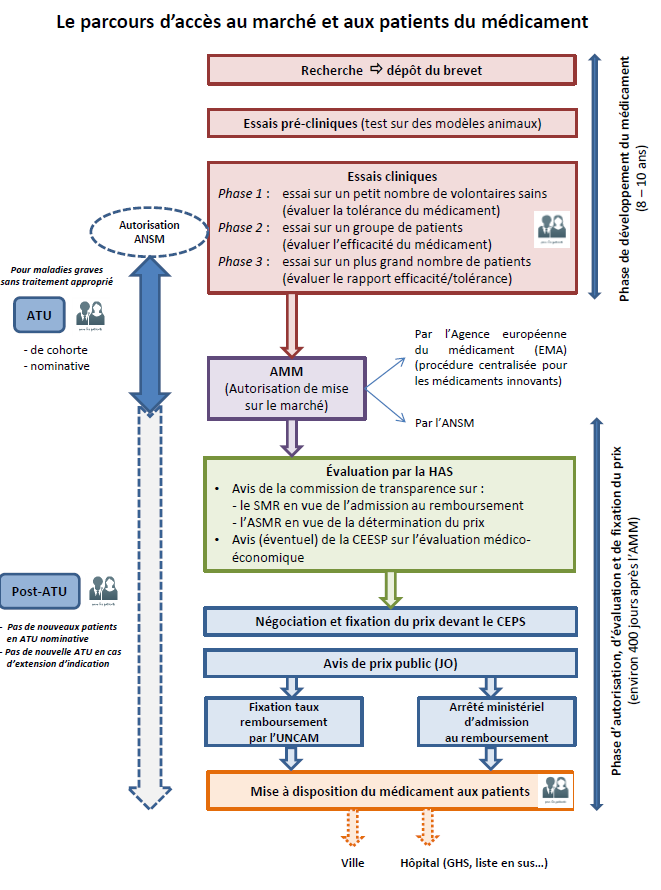
SYNTHÈSE : LE PARCOURS D'ACCÈS AUX MÉDICAMENTS
|
Du médicament au patient : synthèse
de la procédure de mise à disposition
Comme l'illustre le schéma ci-après, les patients français peuvent avoir accès à des molécules nouvelles et susceptibles de constituer un progrès thérapeutique à plusieurs étapes de la chaîne de développement et de mise sur le marché du médicament, explicitées ci-après. Les essais cliniques À la suite de l'identification d'une molécule susceptible de répondre à un besoin médical non pourvu et d'une phase de recherche préclinique en laboratoire suivie d'un dépôt de brevet, la réalisation d'un essai clinique vise à évaluer successivement la tolérance du médicament-candidat sur un petit nombre de volontaires sains (phase I), son efficacité sur des volontaires malades (phase II) et son rapport efficacité/tolérance à plus grande échelle (phase III). Lorsqu'il s'agit d' essais dits « précoces », la tolérance et l'efficacité sont évaluées concomitamment, sur des patients sans alternative thérapeutique, dans une démarche qui peut permettre d'allier recherche et accès aux soins . Si les résultats de l'essai clinique sont convaincants, le promoteur peut engager une procédure en vue de l'éventuelle commercialisation du médicament. La procédure « de droit commun » d'accès au marché des médicaments - L'autorisation de mise sur le marché (AMM) , sollicitée par l'industriel, constitue le point de départ de cette procédure. Celle-ci est accordée sur la base d'une évaluation scientifique de l'efficacité, de la sécurité et du bénéfice/risque du médicament dans la ou les indications pour lesquelles l'AMM est demandée. Elle est délivrée, pour les médicaments innovants, essentiellement au niveau européen (selon une procédure dite centralisée), après avis de l'Agence européenne du médicament (EMA). Dans le cas d'une procédure nationale, c'est en France l'Agence nationale de sécurité du médicament et des produits de santé (ANSM) qui procède à cette évaluation et à l'examen du dossier. - À partir de l'octroi de l'AMM, l'industriel qui commercialise un médicament peut demander son inscription sur la liste des produits remboursables : s'engage alors, au niveau national (et selon des modalités différentes d'un pays européen à l'autre) une procédure en vue de l'inscription du médicament au remboursement et de la fixation de son prix. Elle doit en théorie s'inscrire dans un délai de 180 jours, fixé par une directive européenne. - L'évaluation par la Haute Autorité de santé (HAS), autorité indépendante, en constitue la première étape. Il s'agit dans tous les cas d'une évaluation clinique , à laquelle procède la commission de la transparence de la HAS sur la base du dossier remis par l'industriel. L'objectif est de déterminer : si le produit doit être remboursé et à quel taux, sur la base de son « service médical rendu » (SMR), c'est-à-dire de son intérêt clinique, si le produit apporte un progrès thérapeutique par rapport aux traitements existants, sur la base de l'évaluation de son « amélioration du service médical rendu » (ASMR), qui sert ensuite pour la détermination du prix. Cette évaluation clinique peut être couplée, dans certains cas encore rares, d'une évaluation médico-économique à laquelle procède la commission d'évaluation économique et de santé publique (CEESP) de la HAS. Cette évaluation a vocation à être prise en compte pour la fixation du prix. - La phase de fixation du prix s'engage ensuite sur la base des résultats de l'évaluation, dans le cadre d'une démarche conventionnelle entre les industriels et le Comité économique des produits de santé (CEPS), organisme interministériel. En cas d'accord, cette procédure aboutit à la publication d'un prix public au Journal Officiel, sachant que des remises conventionnelles protégées par le secret des affaires sont négociées entre les industriels et le CEPS (ce prix net de remises n'est pas public). - Ce n'est qu'au terme de ces deux séquences - et après la publication de l'arrêté ministériel d'admission au remboursement du produit et la fixation de son taux de remboursement par le directeur général de l'Uncam (Union nationale des caisses d'assurance maladie) - que le médicament entre dans le circuit de commercialisation « de droit commun » . Il peut alors être distribué en ville (officine) ou dans les établissements de santé selon différents modes de prise en charge : dans le cadre de la rétrocession (médicaments délivrés à l'hôpital mais destinés aux patients en ambulatoire), des GHS (groupes homogènes de séjour), ou encore de la « liste en sus » qui permet la prise en charge de médicaments onéreux en sus des GHS. • Le dispositif des autorisations temporaires d'utilisation (ATU) : une voie d'accès précoce spécifique et dérogatoire La France a prévu que dans certaines conditions, un médicament considéré comme innovant puisse être mis à disposition des patients selon un mode dérogatoire et anticipé, qui se situe en quelque sorte entre les essais cliniques et la commercialisation « de droit commun » : il s'agit d'une autorisation d'utiliser, pour une durée limitée éventuellement renouvelable et « à titre exceptionnel, certains médicaments destinés à traiter des maladies graves ou rares en l'absence de traitement approprié, lorsque la mise en oeuvre du traitement ne peut pas être différée ». Ce dispositif d'autorisation temporaire d'utilisation (ATU) est sollicité soit par l'industriel (ATU de cohorte concernant un groupe de patients dans le cadre d'un protocole déterminé), soit par le médecin prescripteur (ATU nominative pour un patient donné). L'examen de la demande et l'autorisation relèvent de l' ANSM . A ce stade, le produit n'a pas encore fait l'objet d'une évaluation par la HAS ; son prix (appelé indemnité) est par ailleurs fixé librement par le laboratoire . L'ATU est sollicitée en général en fin de période de développement et d'essais cliniques, toujours en amont de l'AMM . Les laboratoires ont l'obligation, quand ils sollicitent une ATU de cohorte, de présenter une demande d'AMM. L'ATU se déroule donc parallèlement à la phase d'examen de l'AMM par l'EMA. Elle prend fin théoriquement au moment de l'obtention de l'AMM . Toutefois, suspendre l'ATU à ce moment, alors que le médicament n'est pas encore distribué car s'engage alors la phase décrite ci-dessus, conduirait à priver les patients, pendant plusieurs mois, de leur traitement. Aussi, une phase dite de « post-ATU », toujours dérogatoire, prend le relais , pendant que se déroule la procédure d'évaluation par la HAS puis celle de fixation du prix. On voit donc que l'intérêt du dispositif d'ATU est non seulement d'anticiper l'AMM mais également, et de plus en plus, d'anticiper la commercialisation effective du médicament selon le circuit de droit commun après son AMM. Les difficultés qui se posent aujourd'hui au dispositif d'ATU, en particulier pour traiter des extensions d'indication d'un médicament sous ATU ou des modalités de régulation financière du dispositif, se comprennent donc à la lumière de l'analyse de cette procédure de « droit commun » dont les délais ont tendance à s'allonger dans un contexte de forte tension sur les prix. |
LISTE DES PROPOSITIONS
_________
Restaurer l'attractivité et l'efficacité du dispositif des autorisations temporaires d'utilisation (ATU) en l'adaptant aux réalités nouvelles de l'innovation
1. Rendre le dispositif des ATU plus rapide et plus souple, mais révisable à tout moment sur la base des données obligatoirement produites au cours des phases d'ATU et de post-ATU
2. Aménager le dispositif de post-ATU nominative pour autoriser les initiations de traitement après la délivrance de l'autorisation de mise sur le marché (AMM)
3. Délivrer les ATU par indication et non plus par produit, de manière à couvrir les situations d'extension d'indication survenant après la délivrance de la première AMM
4. Revenir sur le mode de calcul complexe de la remise rétroactivement versée par les laboratoires au titre de la récupération sur l'indemnité de la phase d'ATU, tel que prévu par l'article 97 de la LFSS pour 2017
Anticiper l'innovation et renouer la confiance entre les acteurs
5. Structurer un cadre pérenne d'échanges pour anticiper sur les innovations à venir susceptibles d'impacter le système de santé
6. Envisager une procédure accélérée d'accès au marché (de type fast-track ) pour des produits innovants fléchés comme prioritaires
7. Refonder les critères de l'évaluation du médicament pour renforcer leur lisibilité et leur compréhension
8. Développer l'évaluation médico-économique des médicaments innovants, en prenant en compte de manière globale les économies qu'ils sont susceptibles de générer pour le système de santé. Intégrer dans ce cadre les spécificités liées aux médicaments orphelins
Fluidifier les procédures pour mieux adapter la grille de lecture aux besoins
9. Envisager, pour des médicaments prometteurs insuffisamment développés, la possibilité d'un remboursement temporaire, conditionné à l'apport de données supplémentaires par l'industriel après l'obtention de l'AMM
10. Expérimenter de nouveaux modes de fixation des prix, plus souples et plus fins (prix par indication ou à l'association, prix plus évolutifs en fonction de l'efficacité en vie réelle, enveloppe globale pluriannuelle par pathologie...)
11. Améliorer le cadre de régulation des prescriptions hors-AMM, sur la base de référentiels nationaux de bon usage, pour sécuriser et rendre plus réactif l'accès aux thérapeutiques adaptées en particulier pour des maladies rares ou certaines situations « de niche »
Garantir l'équité d'accès des patients aux traitements innovants
12. Réformer la liste en sus : revoir les critères d'inscription et de radiation afin de déverrouiller l'accès pour les produits avec une amélioration de service médical rendu (ASMR) IV
13. Responsabiliser en parallèle les prescripteurs en créant à titre expérimental un mécanisme d'intéressement à la juste prescription des produits figurant sur la liste en sus
14. Anticiper les sorties de liste en sus en programmant une baisse des prix voire une revalorisation plus régulière des groupes homogènes de séjour (GHS)
15. Répondre aux enjeux de la médecine personnalisée par une gestion plus dynamique du référentiel des actes innovants hors nomenclature (RIHN) : accélérer et fluidifier l'inscription des actes de biologie innovants à la nomenclature
Consolider le rôle des essais cliniques dans l'accès précoce des patients aux traitements innovants
16. Renforcer les comités de protection des personnes (CPP) et leur expertise :
- Adapter le système du tirage au sort en prévoyant que celui-ci s'applique à un groupe restreint de CPP spécialisés en fonction du domaine concerné par l'essai clinique
- Renforcer le niveau d'expertise des CPP, notamment par la mise en place de formations adaptées et la mise à disposition d'experts en cas de besoin
- Poursuivre l'harmonisation des procédures d'évaluation
- Renforcer les moyens administratifs dont disposent les comités
17. Augmenter les moyens de l'ANSM dédiés à l'instruction des essais cliniques précoces et optimiser la procédure de gestion des essais cliniques
18. Poursuivre la simplification de la convention unique et la généraliser à l'ensemble des établissements
EXPOSÉ GÉNÉRAL
I. CONSOLIDER LES AUTORISATIONS TEMPORAIRES D'UTILISATION (ATU), FORCE DU MODÈLE FRANÇAIS D'ACCÈS PRÉCOCE AUX MÉDICAMENTS INNOVANTS
Si le dispositif des autorisations temporaires d'utilisation (ATU) ne représente que l'une des différentes modalités du système français d'accès précoce à l'innovation, il a acquis, depuis sa mise en place en 1994, une dimension très symbolique , au point de constituer aujourd'hui l'emblème du caractère pionnier de la France en la matière .
Ce dispositif quasiment unique en Europe permet, dès avant la première autorisation de mise sur le marché (AMM), un accès rapide et pris en charge par l'assurance maladie aux innovations médicamenteuses les plus prometteuses, susceptibles de répondre à des besoins jusqu'alors non ou mal couverts. Unanimement salué, décrit comme envié à l'étranger , il constitue indéniablement l'une des forces du modèle français d'accès précoce à l'innovation .
Pour autant, ce modèle qui a fait ses preuves - et dont l'efficacité a récemment été illustrée à l'occasion de l'arrivée sur le marché des traitements contre l'hépatite C, les patients français ayant été les plus nombreux et les plus précocement traités, à rebours du rationnement mis en place dans plusieurs pays européens - connaît aujourd'hui des limites .
Alors que des innovations de rupture particulièrement coûteuses, à l'instar des Car-T cells 3 ( * ) , devraient prochainement arriver sur le marché, il apparaît nécessaire, sans le remettre en cause, de faire évoluer le dispositif pour le consolider .
A. UN DISPOSITIF PLÉBISCITÉ PAR L'ENSEMBLE DES ACTEURS DE LA SANTÉ, QUI A CHANGÉ DE NATURE DEPUIIS SA CRÉATION
Les auditions conduites par vos rapporteurs ont permis de constater un attachement aussi unanime qu'enthousiaste aux ATU.
L'ensemble des acteurs auditionnés, qu'il s'agisse des décideurs ou des régulateurs du système de santé, des industriels, des associations de patients ou encore des soignants, ont décrit le dispositif en des termes particulièrement élogieux : « extraordinaire », « particulièrement organisé et performant », « un cadre exceptionnel », « un dispositif essentiel », « un progrès immense à la fois pour les établissements de santé et les patients » ou encore « une chance inouïe pour l'accès aux traitements ».
Le mécanisme a en effet permis, depuis plus de vingt ans, de mettre un grand nombre de médicaments innovants à la disposition des malades atteints de pathologies graves, souvent mortelles, en situation d'impasse thérapeutique, parfois plus d'un an avant la délivrance de l'AMM - ce qui représente un gain de chances considérable.
Il faut noter toutefois que ce dispositif présente d'autant plus d'intérêt qu'il permet de passer outre les lenteurs de l'accès au marché des médicaments par la voie de droit commun 4 ( * ) .
L'ATU repose sur un socle de principes toujours d'actualité, en dépit d'une série d'évolutions et d'adaptations.
1. Des principes garantissant un accès précoce et universel aux médicaments innovants
a) Un dispositif conçu comme temporaire et dérogatoire, visant à répondre aux situations d'impasse thérapeutique pour les patients atteints de pathologies graves ou rares
Créé par un décret du 8 juillet 1994 5 ( * ) pris sur la base des dispositions de l'article 21 de la loi du 8 décembre 1992 6 ( * ) , le dispositif d'ATU a été conçu comme un mécanisme temporaire et dérogatoire d'accès précoce.
L'objectif en était, au moment de sa création, d'assurer un accès précoce aux nouveaux médicaments anti-VIH.
• Aux termes de l'article L. 5121-12 du code de la santé publique, cette voie d'accès ne peut être mise en oeuvre qu'« à titre exceptionnel », dans les conditions suivantes :
- elle ne peut concerner que des médicaments destinés au traitement de maladies graves ou rares : en pratique, les ATU sont principalement utilisées dans les champs du traitement contre les cancers, le VIH, les hépatites et les maladies rares ;
- elle est réservée aux cas cliniques pour lesquels aucun traitement approprié n'est disponible , c'est-à-dire aux situations d'impasse thérapeutique ;
- elle est ouverte aux patients dont le traitement ne peut être différé ;
- les produits concernés doivent présenter une balance bénéfices/risques supposée favorable à partir de données préliminaires. Les ATU intervenant aux étapes précoces du développement d'un médicament, alors que la phase de recherche est parfois en cours, il n'est pas possible de disposer à ce stade de données cliniques exhaustives et fiables.
• Il existe deux catégories distinctes d'ATU :
- Les ATU de cohorte (ATUc) s'adressent à des groupes de patients traités et surveillés en application de critères définis, le plus souvent, dans un protocole d'utilisation thérapeutique et de recueil d'informations (PUT). Elles sont délivrées à la demande des industriels , qui doivent avoir déposé une demande d'AMM, ou s'engager à le faire dans un délai maximal d'un an à compter de la date d'octroi de l'ATU 7 ( * ) . Elles sont valables pour une durée d'un an renouvelable. Les ATU de cohorte étant généralement délivrées dans un délai très bref avant la délivrance de l'AMM, à un stade avancé du développement clinique du médicament, elles portent le plus souvent sur des produits pour lesquels existe une forte présomption de sécurité et d'efficacité.
- Les ATU nominatives (ATUn), délivrées pour un patient nommément désigné, visent à prendre en compte la situation thérapeutique particulière de patients isolés, qui ne peuvent prendre part à un protocole de recherche biomédicale. En pratique, il s'agit le plus souvent de personnes souffrant de maladies rares. Dans ce cas, l'ATU est délivrée à la demande et sous la responsabilité du médecin prescripteur , dès lors que le médicament est susceptible de présenter un bénéfice pour le patient concerné.
• Quelle que soit la catégorie, l'ATU est autorisée par l'Agence nationale de sécurité du médicament et des produits de santé (ANSM) , au cas par cas s'agissant des ATU nominatives, en tenant compte de l'urgence thérapeutique, et sur la base d'un dossier présenté par l'industriel dans le cas d'ATU de cohorte 8 ( * ) . L'ANSM n'a communiqué à vos rapporteurs aucune donnée relative au délai moyen d'instruction des dossiers. Des représentants de laboratoires ont évoqué des délais très variables, d'environ quatre mois en moyenne.
• Le circuit de distribution des médicaments sous ATU est également dérogatoire . Du fait de l'absence d'AMM, les produits concernés ne sont pas disponibles en officine : ils ne peuvent être prescrits que par des médecins hospitaliers, et sont dispensés par les pharmacies hospitalières. Ils sont ainsi administrés aux patients hospitalisés ou, dans certaines conditions, délivrés au public dans le cadre de la rétrocession hospitalière.
b) Un dispositif permettant un accès large à l'innovation
(1) Les ATU de cohorte permettent de traiter un grand nombre de patients chaque année selon une procédure équitable et transparente
L'ANSM, responsable de la délivrance des ATU, affiche une politique volontariste de développement des ATU de cohorte : « L'ANSM développe depuis 2012 une nouvelle politique dont l'objectif est de privilégier, pour tous les patients en situation d'impasse thérapeutique, un accès équitable et encadré aux traitements innovants, par le développement des ATU de cohorte » 9 ( * ) .
Cette politique correspond à l'une des quatre orientations stratégiques définies dans le contrat d'objectifs et de performance passé entre le ministère de la santé et l'ANSM en juillet 2015 pour la période courant jusqu'en 2018. Celui-ci précise que l'un des objectifs de l'agence est de « favoriser un accès rapide, encadré et large à l'innovation et à l'ensemble des produits de santé pour les patients ».
Les coordonnateurs du huitième conseil stratégique des industries de santé (CSIS) ont reconnu que l'ANSM se montre « plutôt généreuse » lorsqu'il s'agit d'accorder une ATU .
En 2016, 11 909 patients 10 ( * ) ont été inclus dans des ATU de cohorte. Il est à noter que les cohortes sont évolutives et ne sont pas fermées à de nouveaux patients après le premier octroi de l'ATU, ni même après la délivrance de l'AMM : les patients peuvent continuer à bénéficier de l'accès précoce au produit par le biais de l'ATU jusqu'à sa mise à disposition dans le circuit de droit commun.
Évolution du nombre de patients inclus dans les ATU de cohorte

Source : Rapport d'activité de l'ANSM pour 2016
Douze spécialités pharmaceutiques ont été autorisées dans ce cadre en 2016. Au total, la même année, 23 médicaments bénéficiaient d'une ATU de cohorte.
Bilan des ATU de cohorte
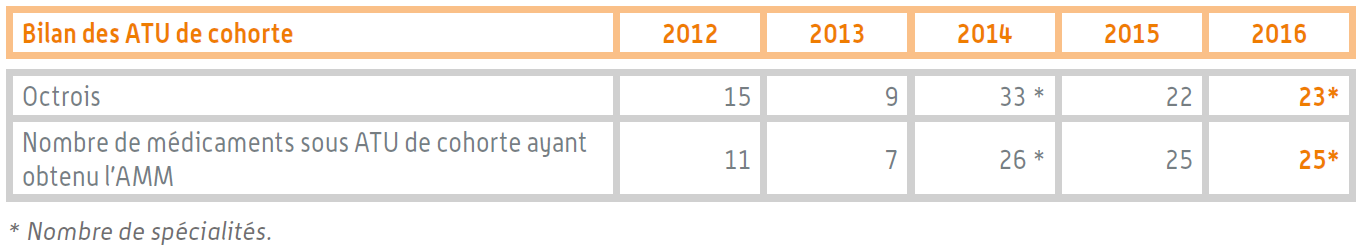
Source : Rapport d'activité de l'ANSM pour 2016
(2) Les ATU nominatives permettent de contourner les aléas de la mise à disposition des produits de santé, liés notamment à la stratégie industrielle des laboratoires
Les ATU nominatives répondent à des enjeux différents dans la chaîne d'accès aux médicaments.
Comme pour les ATU de cohorte, il peut s'agir de permettre un accès rapide à l'innovation avant l'inscription d'un médicament dans le circuit de droit commun ; cet accès se fait cependant au cas par cas. L'accès peut être alors souvent plus précoce encore que dans les ATU de cohorte et intervenir très en amont de la délivrance de l'AMM , alors que des essais cliniques sont toujours en cours.
Certaines ATU nominatives sont octroyées sur le long cours, dès lors que l'industriel ne dépose pas d'AMM pour le produit concerné . L'ANSM a cependant souligné devant vos rapporteurs que de telles situations ne sont pas sans présenter un risque de rupture dans l'accès aux produits concernés, dans le cas où le laboratoire abandonnerait leur production.
L'ordre de grandeur du nombre d'ATU nominatives délivrées est stable au cours des dernières années, de même que le nombre de patients inclus. Au total, un peu plus de 27 000 ATU nominatives ont été délivrées en 2016, portant sur 205 médicaments .
Bilan des ATU nominatives
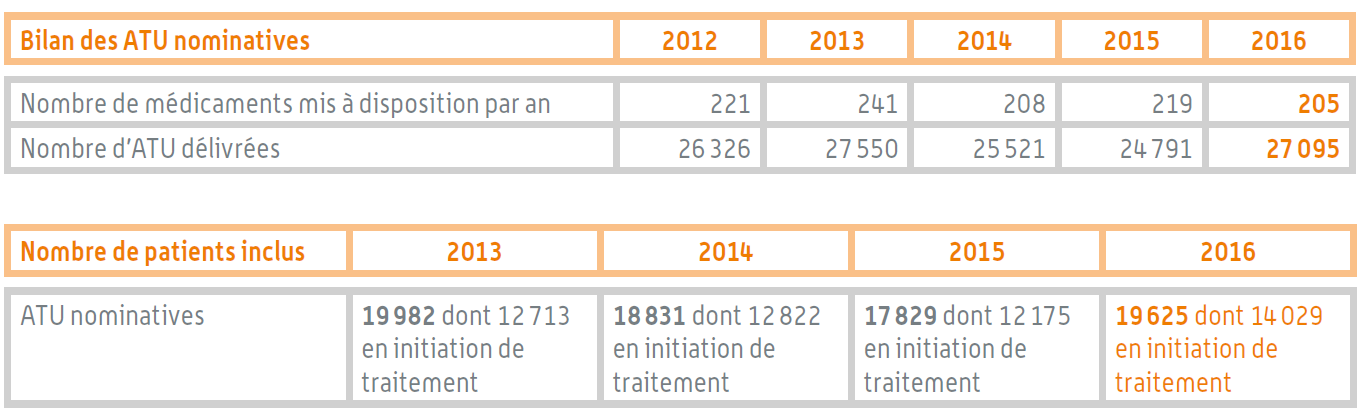
Source : Rapport d'activité de l'ANSM pour 2016
c) Un dispositif financé par l'assurance maladie : un élément d'attractivité indéniable
Tout médicament faisant l'objet d'une ATU est pris en charge par l'assurance maladie dès l'octroi de cette autorisation, sur la base d'un prix libre (appelé « indemnité »), fixé par les laboratoires.
Ce financement est assuré au-delà de l'obtention de l'AMM , jusqu'à l'inscription du produit sur l'une des listes ouvrant droit au remboursement - deux bornes marquant le début et la fin de la période dite de « post-ATU » . Les montants correspondants sont retracés dans les charges du fonds de financement de l'innovation pharmaceutique (FFIP), créé par la loi de financement de la sécurité sociale pour 2017 dans le but de lisser sur plusieurs années le surcroît de dépenses liées aux vagues d'innovations.
Les industriels, à l'instar du Leem, sont unanimes à saluer le dispositif comme « un élément différenciant positif pour l'attractivité de la France ». Il se distingue en effet du mode de financement prévalant pour les essais cliniques, à la charge des laboratoires. En outre, la liberté tarifaire durant cette phase permet aux industriels de fixer des prix élevés, qui servent ensuite de référence pour la négociation des prix dans les autres pays de l'Union européenne.
d) Un dispositif qui reste unique en Europe
• Le dispositif des ATU ne serait plus si spécifique à la France, de l'avis de certaines personnes entendues par vos rapporteurs. Des pays comparables au nôtre ont en effet développé des mécanismes similaires, de sorte que la France continuerait aujourd'hui de bénéficier d'une avancée d'il y a vingt ans, sans préparer un avenir probablement plus concurrentiel.
De nombreux États européens, parmi lesquels l'Allemagne, le Royaume-Uni 11 ( * ) , la Belgique ou encore l'Espagne, ainsi que les États-Unis, ont mis en place des programmes d' early access s'inspirant en plus ou moins grande partie des ATU de cohorte françaises 12 ( * ) .
Ces programmes ne constituent pas nécessairement un dispositif entièrement spécifique au même titre que les ATUc, mais prennent le plus souvent la forme de fast-tracks , c'est-à-dire de procédures accélérées .
En Allemagne, le dispositif de mise à disposition précoce de médicaments non encore autorisés sur le marché est ainsi entré en vigueur le 22 juillet 2010, sur le fondement de l'amendement de la Section 80 du German Medicines Act et dans les suites de l'ordonnance sur les produits médicaux pour usage compassionnel du 14 juillet 2010 (AMHV). Ces textes ont fixé des critères d'éligibilité au dispositif proches de ceux existant en France : le traitement doit cibler un groupe spécifique de patients atteints d'une maladie grave ne pouvant être traités par un médicament autorisé ; une demande d'autorisation de mise sur le marché ou un essai clinique doivent être en cours.
• Ces dispositifs d'accès précoce ne sont cependant pas entièrement comparables au système français des ATU. En Allemagne, la durée du programme est limitée à douze mois. Surtout, leurs modalités de financement diffèrent sensiblement du système français puisque les produits sont mis à disposition à titre gracieux par les laboratoires en Allemagne comme au Royaume-Uni.
Le rapport « Charges et produits » de l'assurance maladie pour 2016 relève en ce sens que les médicaments innovants contre l'hépatite C se sont diffusés beaucoup plus rapidement en France, soulignant que notre pays a donc eu, « par rapport à d'autres pays, une politique volontariste d'accès à ces médicaments nouveaux représentant un réel progrès pour les malades, et ce malgré le coût très élevé des traitements ».
2. Un dispositif qui a changé de nature depuis sa création
Le dispositif des ATU n'est plus, ou plus seulement, un mécanisme compassionnel visant à prendre en compte le profil thérapeutique particulier de certains patients, mais un dispositif structuré d'accès précoce au marché destiné à de grands volumes de patients, dont le coût pour l'assurance maladie représente désormais plus d'un milliard d'euros par an . Cette évolution a connu une forte accélération au cours de la période récente.
a) Une évolution du nombre et de la nature des produits pris en charge
• D'après le rapport à la commission des comptes de la sécurité sociale de septembre 2017, le nombre de produits bénéficiant d'une ATU a connu une forte hausse entre 2013 et 2014 , passant de 206 à 268 médicaments « en stock » dans le dispositif entre 2012 et 2014. Un pic a été atteint en 2015, avec 281 médicaments pris en charge dans ce cadre. Le rapport estime que le nombre de produits bénéficiant du dispositif reste depuis stable autour de 270 prises en charge annuelles.
Cette augmentation concerne principalement les ATU de cohorte , le nombre de médicaments bénéficiant annuellement de ce régime étant passé de 5 à 23 entre 2008 et 2016, selon les services ministériels.
Évolution du nombre d'inscriptions et de sorties
du dispositif ATU
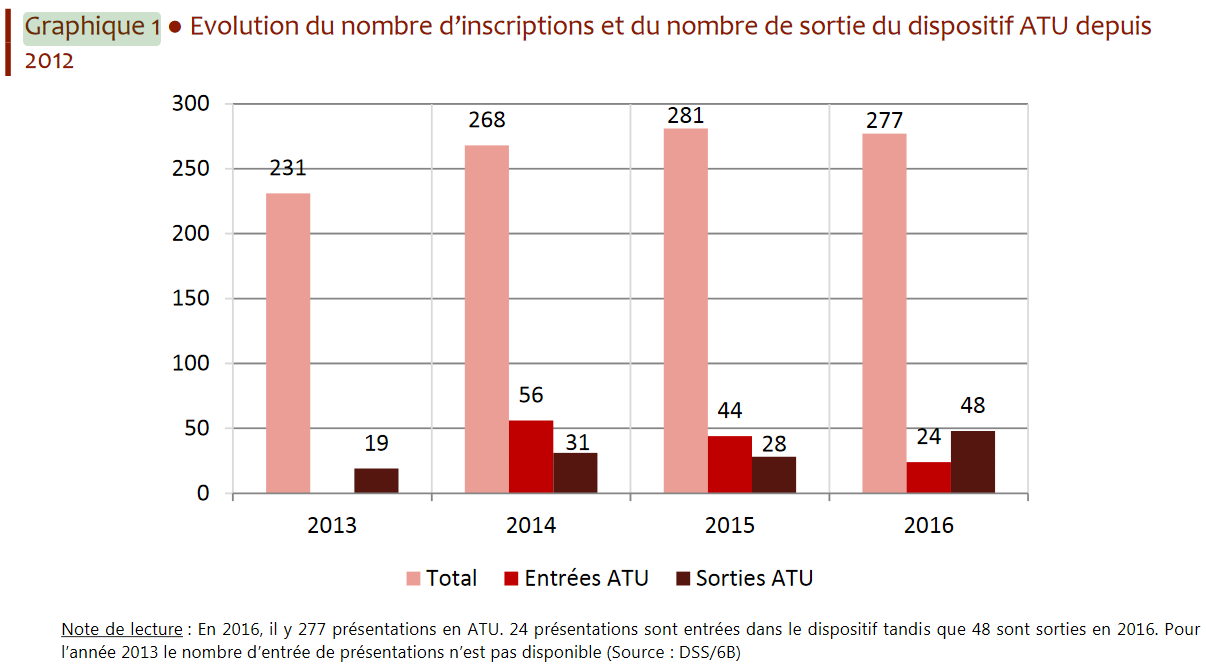
Source : Comptes de la sécurité sociale, rapport de septembre 2017
• La nature des produits bénéficiant d'une ATU de cohorte a également fortement évolué depuis les origines du mécanisme. Alors que le dispositif avait été inspiré, en 1994, par la préoccupation de mettre les produits anti-VIH à disposition des patients, et que les ATU, jusqu'au début des années 2000, portaient principalement sur des médicaments indisponibles en France, les ATU de cohorte délivrées aujourd'hui visent principalement des médicaments anticancéreux avant leur commercialisation en France. Ainsi, sur les douze spécialités pharmaceutiques entrées dans le dispositif des ATUc en 2016, six s'inscrivaient dans le domaine de l'hématologie et de la cancérologie 13 ( * ) .
Le rapport « Charges et produits » de l'assurance maladie pour 2016 relève que le traitement du cancer connaît une profonde mutation , résultant « du développement accéléré des innovations et des bouleversements dans la prise en charge ». Ces évolutions se traduisent par le développement des chimiothérapies orales , qui ont représenté plus de la moitié des nouvelles molécules autorisées entre 2010 et 2014, et la forte progression de la médecine de précision dans les prises en charge oncologiques.
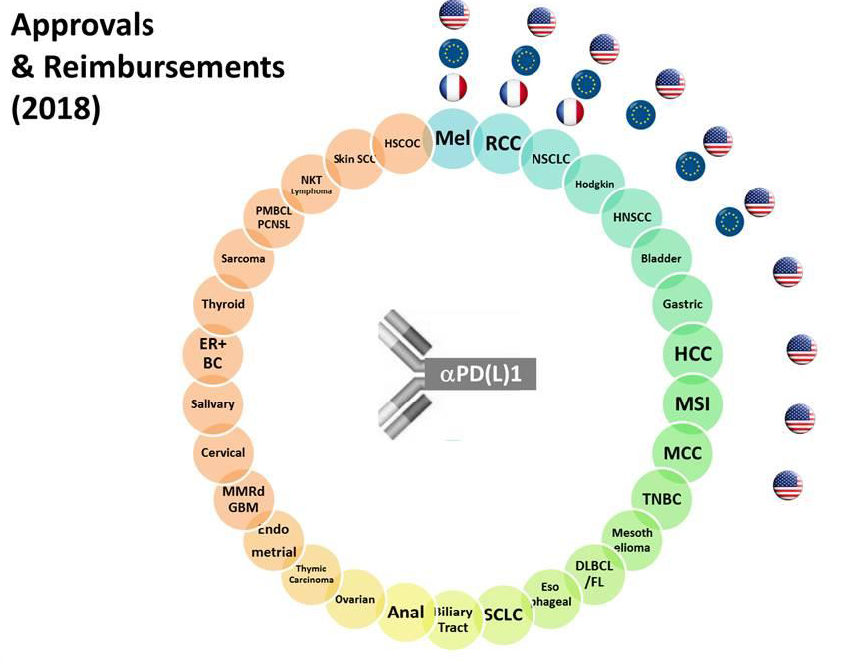
Celle-ci recouvre deux types de traitements : les thérapies ciblées et les immunothérapies spécifiques, dont la part dans le marché des anticancéreux a progressé de 11 à 46 % en dix ans.
|
Les nouvelles approches
thérapeutiques
• La médecine de précision, également appelée médecine personnalisée, a pour objectif de proposer au patient un traitement adapté aux caractéristiques de sa tumeur. Elle repose actuellement sur deux types de traitements : les thérapies ciblées , en premier lieu, sont dirigées contre une voie de signalisation cellulaire plus que contre une tumeur ou une localisation particulière ; l' immunothérapie spécifique, en second lieu, consiste à aider le système immunitaire à reconnaître et s'attaquer lui-même aux cellules cancéreuses. • Selon le Dr Aurélien Marabelle, directeur du programme d'immunothérapie de l'Institut Gustave Roussy, rencontré par vos rapporteurs à l'occasion de leur déplacement, les immunothérapies constituent « un changement radical de paradigme » en oncologie 14 ( * ) . - La première raison est de nature scientifique : ces médicaments ciblent non plus les cellules cancéreuses, mais le système immunitaire du patient : il s'agit d'aider le patient à se défendre lui-même contre le cancer en stimulant son immunité. - La deuxième raison est d'ordre clinique, la prise en charge du patient se trouvant très largement simplifiée (les traitements sont en effet faciles à préparer et à administrer, et ne nécessitent aucune prémédication du patient). Si les bénéfices de ces traitements jusqu'ici mis en évidence sont très importants en termes de survie globale, leurs possibles effets secondaires sont cependant à la fois nouveaux et potentiellement très graves. • Les premiers médicaments de ce type disponibles sur le marché français sont les anticorps dits « anti-PD1 » ou « anti-PDL1 » du fait de leur mode d'action 15 ( * ) . Les Car-T cells 16 ( * ) , dont la stratégie de traitement des cancers a été consacrée « avancée thérapeutique de l'année » en 2017 par la société américaine d'oncologie clinique (2017), devraient prochainement arriver en France, dans un premier temps sous forme d'ATU nominatives. • À ce jour, la médecine de précision ne concerne pas tous les cancers ou tous les patients. Elle permet cependant déjà de développer de nouveaux traitements ciblant précisément des mécanismes biologiques jouant un rôle majeur dans le développement des tumeurs, ainsi que d'identifier des groupes de patients dont les tumeurs présentent des anomalies moléculaires communes et susceptibles d'être ciblées par des traitements spécifiques. |
En particulier, l'arrivée sur le marché des nouveaux traitements prometteurs que constituent notamment les anti-PD1 et prochainement les CAR-T cells constitue un bouleversement majeur pour notre système de santé - en termes à la fois d'équipements, de parcours de soins, de compétences professionnelles et de financement. À titre d'exemple, la progression de ce type de thérapies anticancéreuses entraînerait un fort développement des hospitalisations de jour. Le président de l'Institut national du cancer (INCa) a ainsi indiqué à vos rapporteurs que des travaux, initiés et coordonnés par l'Institut, étaient actuellement conduits dans le but d'anticiper l'arrivée des CAR-T cells .
b) Un dispositif de plus en plus coûteux
Conséquence du dynamisme du nombre d'ATU octroyées et de l'apparition de ces innovations de rupture, peu nombreuses mais au prix extrêmement élevé, le coût du dispositif emprunte une trajectoire d'augmentation très rapide.
Selon les chiffres fournis par la direction de la sécurité sociale, alors que la dépense liée aux ATU plafonnait à 110 millions d'euros annuels jusqu'en 2013, elle a atteint deux pics successifs à un milliard d'euros en 2014 et 2016.
Évolution de la dépense au titre des
dispositifs d'ATU
et de post-ATU pour les années 2014 à
2016
|
2014 |
982 millions d'euros, dont : - 47 millions au titre des médicaments délivrés en hospitalisation
- 935 millions en ambulatoire
via
la
rétrocession, dont 893 millions
|
|
2015 |
673 millions d'euros, dont : - 144 millions en hospitalisation, dont 84 millions au titre des anti-PD1
- 527 millions d'euros en ambulatoire
via
la
rétrocession,
|
|
2016 |
997 millions d'euros, dont : - 471 millions en hospitalisation, dont 318 millions au titre des anti-PD1 - 526 millions d'euros en ambulatoire via la rétrocession. |
Source : Ministère des solidarités et de la santé
• Ces deux chocs successifs sur les dépenses d'ATU sont liés à l'arrivée des nouvelles molécules innovantes précitées :
- le premier, en 2014 et 2015, résultait de l'arrivée sur le marché des nouveaux traitements contre l'hépatite C , dits antiviraux à action directe (AAD) 17 ( * ) . Ces dépenses sont désormais et depuis 2016 prises en charge dans le cadre du droit commun du remboursement des produits de santé ;
- le second, en 2016 (dernière année pour laquelle les données sont disponibles), résulte de l'arrivée dans le circuit hospitalier des anti-PD1 , nouvelles molécules anticancéreuses particulièrement onéreuses. Deux molécules principalement contributrices à cette dépense, Opdivo® (210 millions d'euros en 2016) et Keytruda® (61 millions d'euros pour la même année), sont sorties du dispositif ATU et post-ATU en janvier 2017 pour leurs principales indications 18 ( * ) .
En 2015, les « anti-PD1 » et les médicaments contre l'hépatite C ont représenté 75 % du coût total des médicaments sous ATU.
• Ces surcroîts de dépenses s'entendent compte tenu des efforts déployés par les pouvoirs publics pour en contenir l'ampleur.
Le surcoût lié à l'arrivée des nouveaux traitements contre l'hépatite C a ainsi été limité par la mise en place d'un mécanisme de régulation spécifique introduit par la loi de financement de la sécurité sociale pour 2015, dit « enveloppe W » 19 ( * ) .
L'assurance maladie a lancé parallèlement, dès janvier 2015, une campagne d'accompagnement auprès des établissements rétrocédant ces médicaments, dans le but de promouvoir leur bon usage.
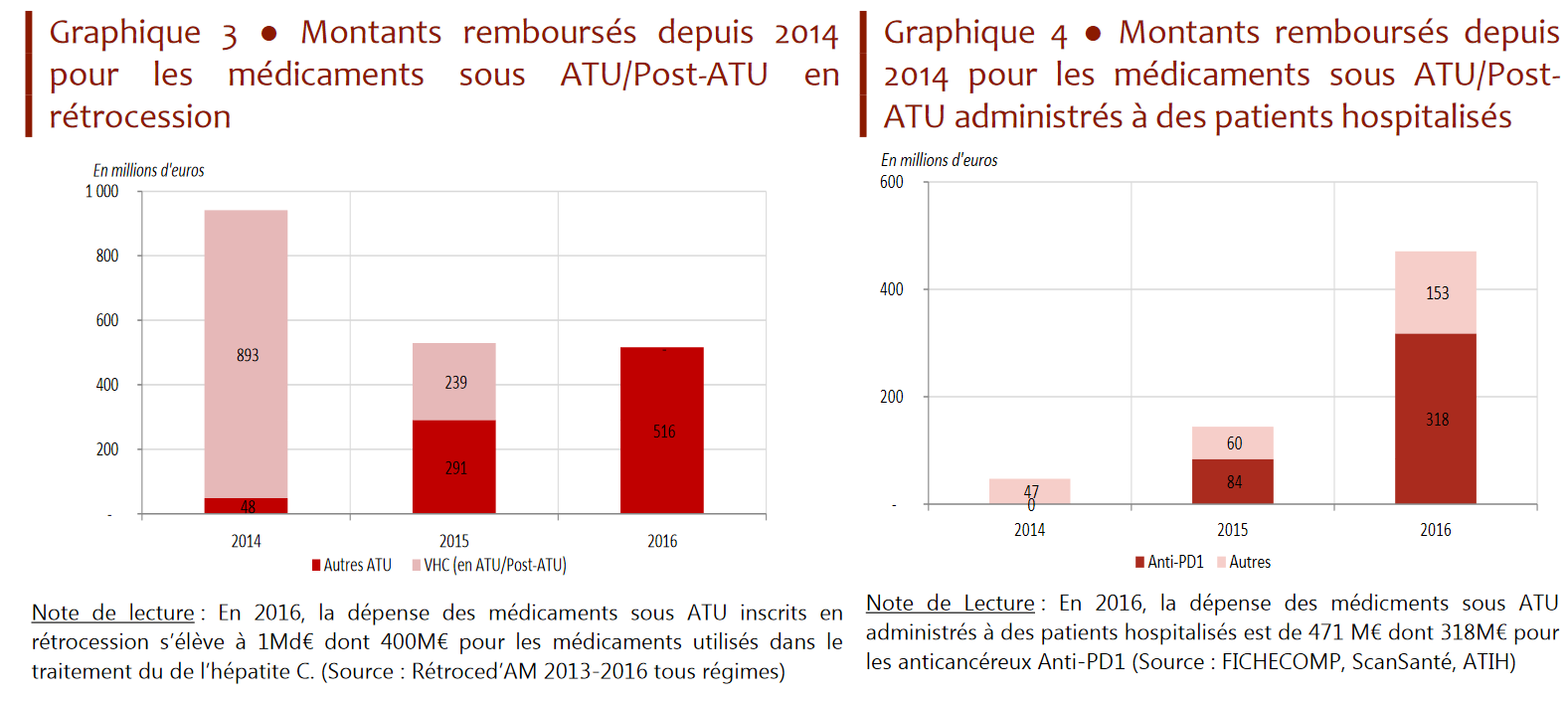
Source : Comptes de la sécurité sociale, rapport de septembre 2017
En somme, le poids des dépenses ambulatoires liées aux médicaments anti-VHC a été remplacé, au tournant de l'année 2015, par celui des anti-PD1, en forte croissance, dans les dépenses hospitalières. Les dépenses d'ATU sont ainsi très concentrées sur quelques médicaments.
En 2016, près de 80 % de la dépense hospitalière au titre des ATU porte ainsi sur quatre produits anticancéreux , parmi lesquels les deux anti-PD1 précités (Opdivo® et Keytruda®).
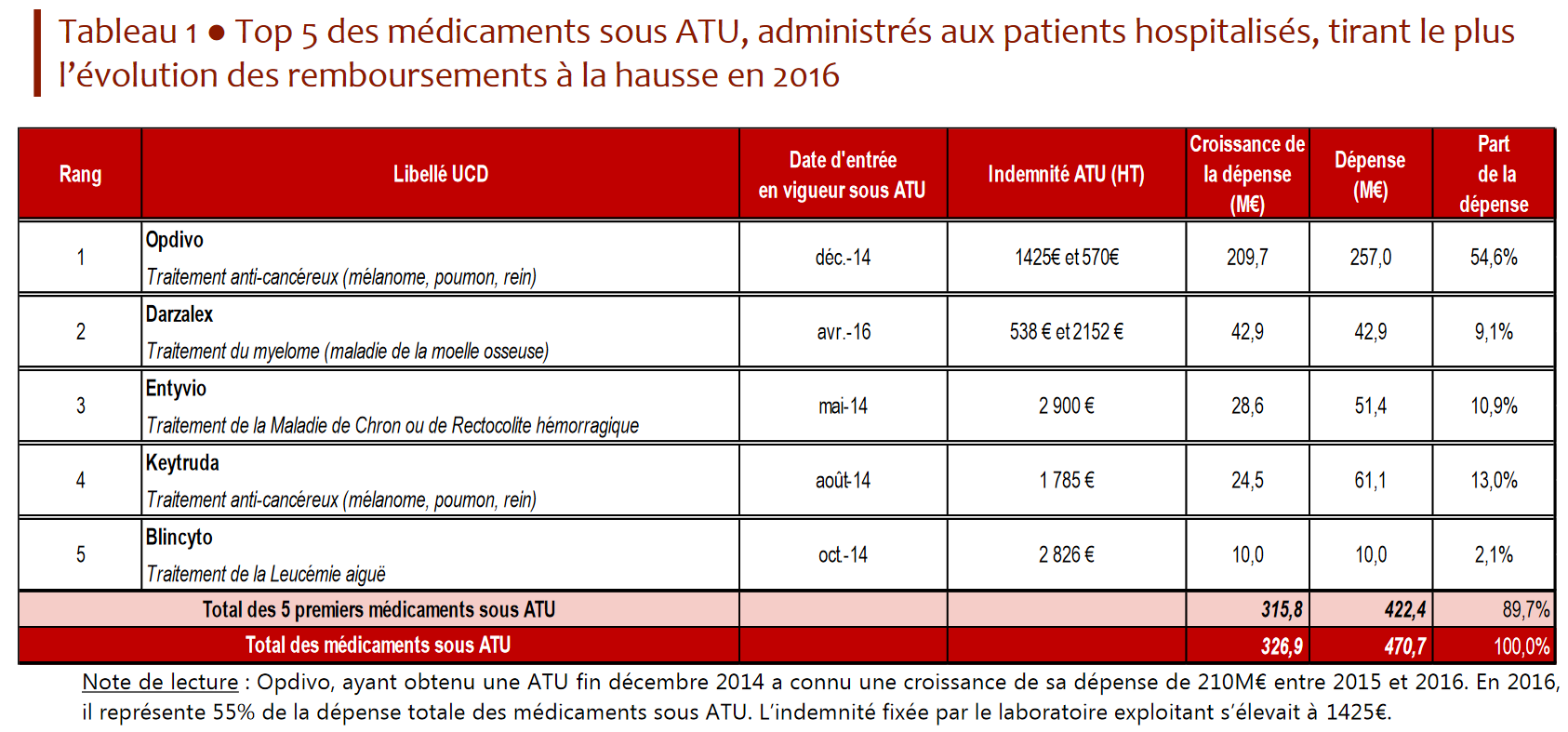
Source : Comptes de la sécurité sociale, rapport de septembre 2017
• Cette « escalade des coûts » représente un défi pour la soutenabilité, à terme, des ATU. Cette question n'est d'ailleurs ni propre au dispositif des ATU, ni propre à la France.
Elle se pose toutefois de manière particulièrement vive dans le cadre des ATU, dans la mesure où l'indemnité de mise à disposition est fixée par les laboratoires, sans négociation avec les pouvoirs publics.
Pour de nombreux interlocuteurs, l'impact budgétaire prévisible des nouveaux traitements contre le cancer est susceptible de remettre en cause la capacité du système de protection sociale français à maintenir un accès à l'innovation et aux meilleurs traitements pour tous les patients.
Dans son récent rapport sur le prix des médicaments 20 ( * ) , l'INCa relevait ainsi que « des innovations en cancérologie sont annoncées par rafales sur le marché dans le monde et en France . Elles devraient constituer une rupture dans l'attitude générale vis-à-vis des cancers en commençant par les formes avancées. Ces innovations parfois très coûteuses, aujourd'hui les immunothérapies spécifiques médicamenteuses et demain les CAR-T, vont mettre à rude épreuve la soutenabilité financière de ce poste de dépenses ».
Alors que 3,2 milliards d'euros de dépenses sont aujourd'hui consacrés aux médicaments anticancéreux, le surcoût attendu des nouveaux traitements est estimé entre 1 et 1,2 milliard par l'observatoire du cancer de l'Institut Curie 21 ( * ) - au-delà du strict cadre des ATU. L'observatoire indique par ailleurs qu'une thérapie ciblée contre le cancer coûte aux alentours de 50 000 euros par an et par patient, soit 5 à 10 fois plus qu'une chimiothérapie classique ; le coût estimé pour une immunothérapie atteindrait quant à lui au moins 80 000 euros par an et par patient.
Les défis ne seront par ailleurs pas limités au seul développement quantitatif des innovations médicamenteuses. Les modes de prises en charge ainsi que les stratégies thérapeutiques recourant à ces nouvelles molécules devraient dans le même temps se diversifier , faisant intervenir des associations de thérapies ciblées ou une utilisation séquentielle de ces thérapies, ce qui contribuera à diffuser largement le recours à ces traitements. Ceux-ci supposent de nouvelles formes de prise en charge qui pourraient donner lieu à des évolutions profondes de l'organisation de notre système de soins.
Vos rapporteurs ont par ailleurs relevé, à chacune de leurs rencontres avec des professionnels de santé, que la progression des prétentions tarifaires des laboratoires entretient une forte inquiétude des praticiens de première ligne quant à une possible dégradation de la prise en charge des patients. En témoignent les prises de position publiquement affichées par des médecins cancérologues, en France comme à l'étranger, notamment sous forme de tribunes dans la presse. En 2016, cette question a fait l'objet d'un débat en assemblée plénière dans le cadre de l' American society of clinical oncology (Asco), le plus grand congrès international en matière d'oncologie, qui fait figure de référence en ce domaine.
Certains interlocuteurs ont considéré que le système actuel favorise à certains égards le maintien de prix élevés sur le long terme : les médicaments extrêmement coûteux arrivant à l'hôpital sous ATU sont le plus souvent inscrits sur la liste en sus après l'obtention de l'AMM, et continuent ainsi à bénéficier d'une forte indemnisation pendant plusieurs années.
c) Un changement de positionnement dans la chaîne d'accès à l'innovation : l'allongement de la phase de post-ATU
• L'allongement des délais entre l'obtention de l'AMM et la fixation du prix des médicaments a entraîné des problèmes de coordination entre la fin du dispositif d'ATU , marquée par la délivrance de l'AMM, et l'entrée des médicaments pris en charge dans le cadre de droit commun du remboursement des produits de santé.
Se posaient notamment la question de la continuité des traitements entamés par les patients ainsi que celle de l'accès de nouveaux patients aux produits concernés au cours de cette période de transition.
• Afin de répondre à ces difficultés, deux dispositions législatives ont successivement été introduites :
- l'article 24 de la loi dite « Médicament » de 2011 22 ( * ) a d'abord mis en place un dispositif expérimental visant à poursuivre la délivrance et la prise en charge des médicaments ayant obtenu une ATU avant leur inscription au remboursement, pour une période maximale de sept mois ;
- l'article 48 de la loi de financement de la sécurité sociale pour 2014 23 ( * ) a ensuite pérennisé ce dispositif modulo quelques ajustements 24 ( * ) .
• Le dispositif depuis lors en vigueur, qualifié « d'avancée certaine » par plusieurs interlocuteurs, consiste en un régime relais dit de « post-ATU » . Ce régime est ouvert à compter de la date d'arrêt de l'ATU fixée par l'ANSM au moment où le produit concerné obtient une AMM ; il prend fin à compter de son inscription au remboursement.
Pendant cette période, tout médicament qui, préalablement à l'obtention de son AMM, a bénéficié d'une ATU, peut continuer à être délivré par les établissements de santé et pris en charge par l'assurance maladie , jusqu'à la décision d'inscription du produit sur la liste des produits remboursables ; elle est toujours fondée sur le montant de l'indemnité librement fixée par les laboratoires pharmaceutiques.
Le circuit des médicaments bénéficiant d'une ATU
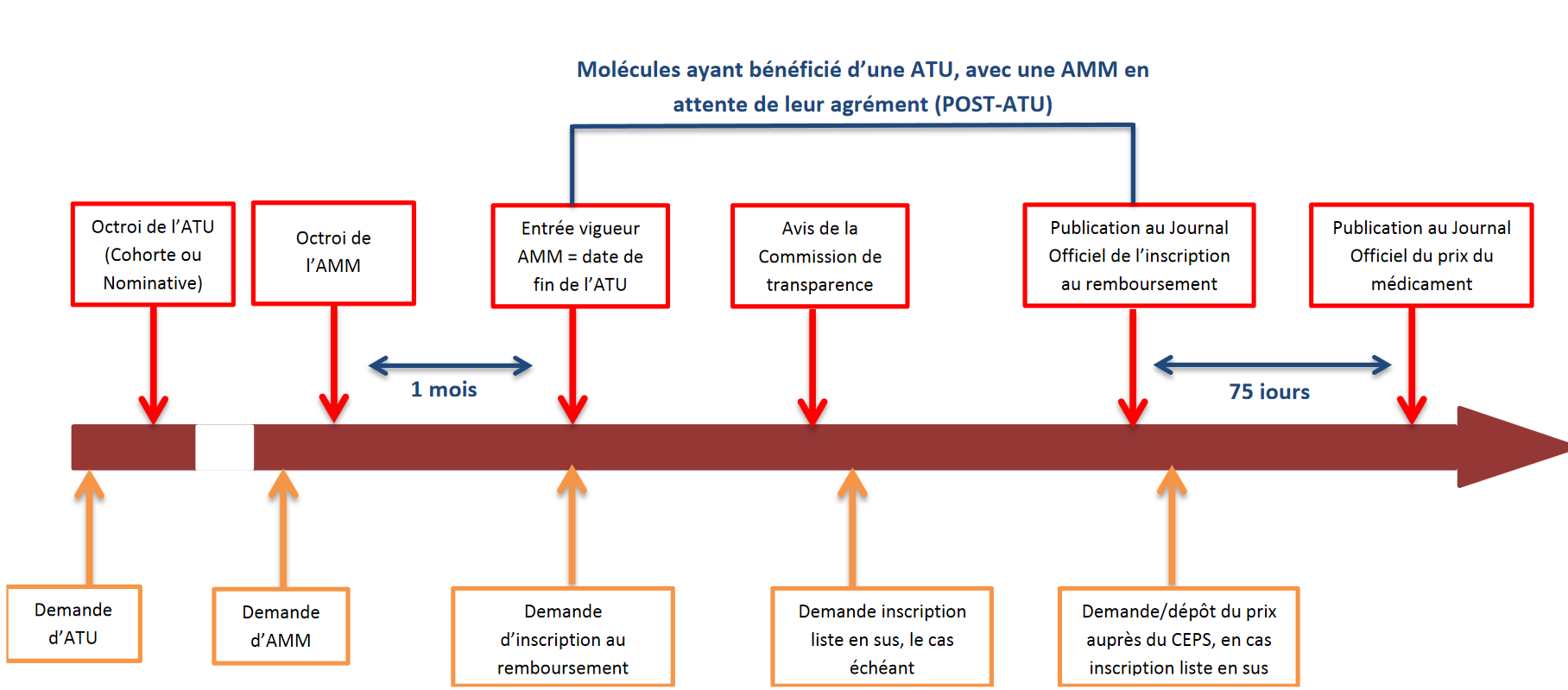
Source : Comptes de la sécurité sociale, rapport de septembre 2017
L'ensemble de ces dispositions visent à éviter toute rupture de prise en charge pour les patients . Que la spécialité ait fait l'objet d'une ATU nominative ou d'une ATU de cohorte, la prise en charge du médicament est garantie pour les patients dont le traitement a été initié sous le régime de l'ATU - à l'exception des cas dans lesquels l'indication considérée fait l'objet d'une évaluation défavorable au titre de son AMM, ou lorsqu'une alternative thérapeutique a pu être trouvée.
• La situation est cependant différente s'agissant des initiations de traitement en post-ATU :
- pour les médicaments ayant fait l'objet d' ATU nominatives , l'initiation de nouveaux traitements en post-ATU n'est pas prise en charge par l'assurance maladie ;
- pour les médicaments ayant fait l'objet d'une ATU de cohorte , sont remboursés les nouveaux traitements s'inscrivant dans les indications de l'ATU, à condition toutefois qu'elles soient mentionnées dans l'AMM ou dans une extension d'AMM en cours d'évaluation. L'initiation de nouveaux traitements est également possible dans des indications mentionnées dans l'AMM mais n'ayant pas fait l'objet de l'ATU de cohorte.
Modalités de prise en charge par l'assurance
maladie des médicaments
en ATU et en post-ATU
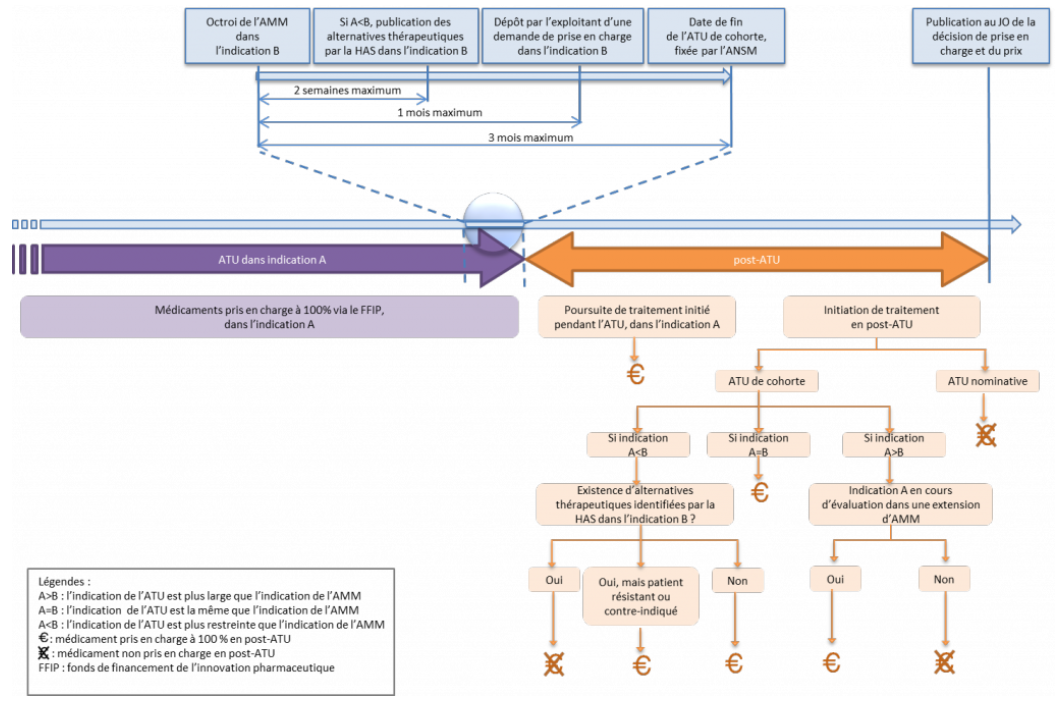
Source : Ministère des solidarités et de la santé
• Selon les données transmises par les services ministériels, la durée moyenne d'une ATU de cohorte est de sept mois . Cette moyenne marque cependant de fortes disparités selon les produits considérés : tandis que certaines sont clôturées au bout de deux ou trois mois, d'autres peuvent durer plusieurs années.
La situation est différente pour les ATU nominatives, pour la durée desquelles il n'est pas possible de dégager une moyenne. Tandis que certaines ATUn visent à autoriser une injection unique d'un produit pour un seul patient, d'autres sont accordées pour le traitement de maladies chroniques, qui peuvent dès lors conduire à un traitement à vie du patient.
La durée de la phase de post-ATU dépend principalement de celle de la procédure d'évaluation et de négociation du prix du médicament en vue de son inscription au remboursement 25 ( * ) .
• Comme cela a été souligné à vos rapporteurs lors des auditions, l'on assiste depuis quelques années, en raison de la plus grande précocité des AMM délivrées, à une compression de la phase d'ATU proprement dite (soit avant la délivrance de l'AMM) et à une extension concomitante de la séquence post-ATU . C'est donc sur cette phase de post-ATU que se concentrent aujourd'hui de nombreux enjeux .
La précocité des AMM conduit par ailleurs à ce que certains médicaments présentant pourtant le « profil » adéquat pour en bénéficier ne s'inscrivent pas dans le dispositif des ATU, faute de temps.
Or, l'ATU, en ce qu'elle permet une première mise à disposition effective des produits aux patients, constitue un atout précieux pour anticiper les conditions d'utilisation du produit en vie réelle, et par voie de conséquences apprécier ses équilibres tarifaires.
B. PRÉSERVER L'ATTRACTIVITÉ ET L'EFFICACITÉ DU SYSTÈME DES ATU EN L'ADAPTANT AUX RÉALITÉS NOUVELLES DE L'INNOVATION
Les auditions conduites par vos rapporteurs ont fait apparaître plusieurs points de crispation dans le mode de fonctionnement actuel du dispositif des ATU , qui ne serait aujourd'hui plus entièrement adapté aux nouveaux modèles de développement et de mise sur le marché des innovations médicamenteuses.
Deux difficultés concentrent les critiques : la question des extensions d'indication de médicaments bénéficiant ou ayant déjà bénéficié d'une ATU, que le dispositif actuel ne permet pas de prendre en charge de manière satisfaisante, et celle de la régulation financière du dispositif ; cette dernière a fait l'objet d'ajustements récents qui se heurtent au modèle économique des entreprises de biotechnologies, devenues des acteurs importants du développement de molécules innovantes.
Au-delà de ces deux sujets, d'autres ajustements apparaissent aujourd'hui nécessaires afin de préserver la pérennité, l'excellence et l'attractivité du dispositif des ATU, et ainsi de garantir un accès rapide, systématique et universel des patients français aux innovations.
1. Améliorer le suivi du dispositif pour en accroître la réactivité
a) Avec le recul, une évaluation hétérogène des produits sous ATU, reflet d'une prise de risque inhérente au dispositif
• L'ensemble des médicaments entrant dans le cadre des ATU ne constituent pas nécessairement des ruptures d'innovation si l'on considère, a posteriori , les résultats de leur évaluation par la Haute Autorité de santé.
Le rapport de septembre 2017 à la commission des comptes de la sécurité sociale indique à cet égard que si « cette évaluation confirme le plus souvent l'efficacité globale des traitements », « leur performance par rapport aux alternatives thérapeutiques, lorsqu'elles existent, n'est pas toujours avérée » 26 ( * ) .
Ainsi, 77 % des molécules admises en ATU depuis 2013 présentent un service médical rendu (SMR) important, 7 % un SMR modéré, 2 % un SMR faible et 5 % un SMR insuffisant et donc ne justifiant pas leur inscription au remboursement.
S'agissant du critère de l'amélioration du service médical rendu (ASMR), seules un peu plus d'un quart des spécialités pharmaceutiques mises à disposition sous ATU ont obtenu un ASMR II ou III , qui signent une amélioration thérapeutique importante ou modérée (2 % une ASMR II et 26 % une ASMR III). La grande majorité des produits (58 %) ont obtenu une ASMR IV, soit une amélioration thérapeutique mineure , et 14 % d'entre eux une ASMR V, soit aucune amélioration thérapeutique par rapport aux comparateurs existants.
• Vos rapporteurs soulignent que ces résultats mitigés doivent être lus avec la nuance qui s'impose, en prenant en compte deux aspects.
En premier lieu, ils sont malgré tout meilleurs que ceux obtenus par l'ensemble des médicaments évalués (ou réévalués) par la commission de la transparence de la HAS 27 ( * ) . Par ailleurs, pour la plupart des médicaments ayant obtenu une ATU, il n'existe en réalité et par définition aucune alternative thérapeutique satisfaisante.
Ainsi, ces résultats sont interprétés de manière divergente par les différents interlocuteurs entendus par vos rapporteurs.
Des fédérations hospitalières ont pu voir, sur ce point comme sur l'augmentation du nombre de produits pris en charge sous ATU, le signe d'une « dérive » dans l'application des critères prévus par l'article L. 5121-12 du code de la santé publique. Autrefois réservées à la prise en charge de pathologies mortelles pour lesquelles il n'existait aucun traitement, les ATU seraient aujourd'hui accordées de manière très large avant de déboucher sur des évaluations médiocres et souvent décevantes.
Le Leem estime pour sa part que le fait que la majorité des produits ayant bénéficié d'une ATU finissent par obtenir une ASMR de niveau IV ou V témoigne de la faible lisibilité du système d'évaluation des médicaments, ainsi que de son absence de prévisibilité.
L'ANSM relève quant à elle que l'hétérogénéité relative qui apparaît dans l'évaluation a posteriori des produits sous ATU est le reflet d'une indispensable politique de prise de risque endossée par l'agence .
Un « verrouillage » en amont du dispositif n'est pas souhaitable ; ce serait contraire à sa vocation de permettre un accès précoce des patients à des traitements prometteurs. En revanche, ces constats plaident pour un meilleur suivi en aval des produits sous ATU.
b) Renforcer le suivi des produits bénéficiant d'une ATU
• Dans la mesure où l'on se trouve, au stade de l'octroi de l'ATU, dans un contexte de simple présomption d'un rapport bénéfices/risques favorable, le recueil et l'exploitation des données cliniques au cours de la mise en oeuvre de l'autorisation constituent des enjeux majeurs pour la bonne évaluation finale des produits supposés innovants.
Ainsi que l'a souligné la présidente de la HAS, le déploiement des ATU constitue en effet une première utilisation des médicaments en vie réelle , qui plus est à grande échelle. Alors que les dossiers d'AMM ne recensent que quelques dizaines ou centaines de situations d'utilisation des produits évalués, les ATU peuvent concerner des milliers de patients.
La phase d'ATU devrait dès lors être l'occasion de conforter l'efficacité et la sécurité des produits pressentis comme innovants, mais aussi d'analyser les pratiques afin de développer leur efficience .
Les représentants de l'INCa ont par ailleurs souligné que la phase d'ATU offre une occasion précieuse de recueil de données intermédiaires entre les informations obtenues au cours des essais cliniques, qui portent sur des patients très sélectionnés, et celles ressortant de l'utilisation en vie réelle, qui se caractérise par une prescription large et donc parfois non totalement pertinente.
• Or, la plupart des interlocuteurs entendus par vos rapporteurs se sont accordés à considérer comme insuffisant le suivi des données d'utilisation, de sécurité et d'efficacité au stade de l'ATU . En dépit des obligations 28 ( * ) qui pèsent sur les professionnels de santé et les laboratoires, le suivi des patients traités dans le cadre des ATU (de cohorte comme nominatives) reste lacunaire ou de médiocre qualité.
Le respect du protocole d'utilisation thérapeutique et de recueil d'informations (PUT) 29 ( * ) , transmis à l'ANSM et auquel la délivrance des ATU de cohorte et de certaines ATU nominatives est pourtant conditionné, n'est par ailleurs pas suffisamment sanctionné.
Les services ministériels ont ainsi estimé que « la visibilité sur les résultats des données d'efficacité et de tolérance, dont le recueil est pourtant intangible à l'octroi d'une ATU, peut faire défaut ».
La commission de la transparence de la HAS déplore quant à elle que les résultats du suivi des médicaments sous ATU ne soient pas de qualité suffisante pour lui permettre d'enrichir son évaluation , notamment lorsque les médicaments sont indiqués dans le traitement de maladies rares, pour lesquels de telles données sont particulièrement précieuses.
|
Fonctionnement et utilisation des protocoles
Dans le cadre des ATU de cohorte et pour certaines ATU nominatives, le recueil des données d'utilisation est obligatoire dans le cadre des protocoles d'utilisation thérapeutique et de recueil d'information. Le PUT est établi entre l'ANSM et le titulaire des droits d'exploitation du médicament. Il permet de fixer les modalités de suivi des patients traités, le recueil de données portant sur l'efficacité, les effets indésirables, les conditions réelles d'utilisation, les caractéristiques de la population bénéficiant du médicament autorisé. Ces données de suivi sont régulièrement soumises à l'autorité compétente (ANSM) tout au long de l'ATU et du post-ATU. Les données issues des PUT sont également soumises dans le dossier de demande déposé auprès de la commission de la transparence de la HAS. Source : ANSM |
• Selon un rapport récemment remis à la ministre en charge de la santé sur les données en vie réelle pour les médicaments 30 ( * ) , ces difficultés s'expliquent en partie par des obstacles techniques apparaissant, à première vue, faciles à résoudre.
Ce rapport relève ainsi que « les suivis d'efficacité et de sécurité [prévus dans le cadre des ATU] s'effectuent dans des conditions qui ne permettent pas d'optimiser l'exploitation des données recueillies ». Le formulaire rempli, dans le cadre des ATU nominatives, par le médecin prescripteur pour obtenir la délivrance initiale puis le renouvellement de l'autorisation, est en effet renseigné manuellement. Les données de suivi collectées dans ce cadre pourraient donc très facilement être mieux valorisées grâce à la simple informatisation de leur recueil .
Le rapport relève à cet égard que dans les exemples étrangers, « cette informatisation conditionne l'exploitation effective des registres, et permet aussi d'impliquer les prescripteurs grâce aux retours d'information qu'elle permet ».
Les représentants du Leem ont néanmoins relevé que le raccourcissement de la phase d'ATU ne permet pas toujours le traitement d'un nombre suffisant de patients pour pouvoir générer des données robustes entre l'octroi de l'ATU et la demande d'AMM.
• Pour autant, vos rapporteurs estiment qu'il n'est pas de bonne gestion de se priver de données aussi précieuses pour l'évaluation des produits de santé, qui plus est alors qu'elles sont déjà produites.
Alors que les accès précoces aux médicaments devraient se développer au cours des prochaines années, sous l'effet de la poursuite du mouvement d'innovation, ces informations apparaissent indispensables à la réforme et à la consolidation de notre système d'évaluation.
Le recueil et l'exploitation de ces informations doivent dès lors être rendus véritablement effectifs .
Une telle évolution pourrait notamment passer par le renforcement des obligations pesant sur les laboratoires s'agissant de la périodicité des transmissions , tout au long de la période d'ATU comme de la séquence de post-ATU. Ces données pourraient par ailleurs être directement transmises à la HAS, et non pas seulement à l'ANSM.
Cela pourrait conduire à rendre les ATU révisables à tout moment , dès lors que l'exploitation des informations transmises n'a pas permis de mettre en évidence un progrès thérapeutique compatible avec le cadre particulièrement favorable des ATU. En contrepartie, la délivrance des ATU pourrait être rendue plus rapide et plus souple , de manière à les faire intervenir davantage en amont de l'octroi de l'AMM.
Un tel système permettrait de mettre fin aux configurations dans lesquelles un produit dont l'évaluation par la HAS ne met finalement pas en avant un progrès thérapeutique a précédemment bénéficié du cadre généreux de l'ATU et du post-ATU pendant, parfois, plusieurs années. Cette situation n'est en effet satisfaisante ni pour les industriels, qui se voient alors contraints de rembourser des montants importants correspondant à la différence entre l'indemnité ATU et le prix finalement fixé par le CEPS, ni pour les pouvoirs publics, qui versent des indemnités élevées ne correspondant pas toujours au service thérapeutique rendu aux patients.
|
Proposition n° 1 : Rendre le dispositif des ATU plus rapide et plus souple, mais révisable à tout moment sur la base des données obligatoirement produites au cours des phases d'ATU et de post-ATU. |
2. Répondre aux situations de rupture d'équité entre patients : améliorer la continuité des prises en charge
Selon les industriels, si les ATU permettent aux patients français de bénéficier d'un accès aux médicaments innovants avant leurs voisins allemands ou anglais, cet accès précoce ne concerne cependant que 10 % de la population cible des produits , en raison de l'étroitesse des critères définis pour la délivrance de l'ATU.
Au-delà de ce constat général, les auditions conduites par vos rapporteurs ont permis de mettre en évidence deux situations problématiques pour l'accès aux soins dans le cadre des ATU , relayées par les professionnels de santé.
• En premier lieu un défaut de formation et d'information des professionnels de santé ainsi qu'un défaut d'information des patients , seraient à l'origine de ruptures d'égalité selon les territoires et les établissements de santé.
France Assos santé considère ainsi que « l'accès équitable aux ATU est rarement constaté dans la pratique ».
La Fehap a souligné par ailleurs que ce défaut d'information des équipes soignantes pouvait être à l'origine de défaillances dans le lien entre le centre de référence dans lequel est traitée la pathologie faisant l'objet de l'ATU et le centre hospitalier dans lequel est habituellement suivi le patient. Elle relève que « les mécanismes d'ATU mériteraient de faire l'objet d'une synthèse pédagogique adressée aux professionnels de santé ».
• Se pose en second lieu le problème de l'entrée dans le régime de post-ATU , dont les critères d'accès ne permettent pas de couvrir la totalité de la population potentiellement bénéficiaire du médicament, alors même que ce régime a tendance à s'étirer dans le temps du fait de l'allongement de la procédure d'accès au marché de droit commun.
Ce problème d'accès aux soins se pose notamment pour les médicaments ayant fait l'objet d'ATU nominatives , dont la prise en charge en post-ATU est limitée aux seules poursuites de traitements engagés sous le régime de l'ATU. Selon les services ministériels, cette disposition avait été initialement introduite dans le but d'inciter les laboratoires à solliciter des ATU de cohorte qui permettent un accès plus transparent aux médicaments et les engagent plus formellement à mettre en place un suivi des patients traités.
Or, vos rapporteurs regrettent que cette limitation de la prise en charge retarde parfois durablement l'accès de patients atteints de maladies graves ou rares à des traitements appropriés . Un aménagement du dispositif de post-ATU pour les produits ayant fait l'objet d'ATU nominatives, de manière à permettre les initiations de traitement pendant cette période, permettrait d'assurer une plus grande équité entre patients.
|
Proposition n° 2 : Aménager le dispositif de post-ATU nominative pour autoriser les initiations de traitement après la délivrance de l'AMM. |
3. Les extensions d'indication, « trou dans la raquette » du dispositif : remédier à un besoin thérapeutique urgent
La question qui fait l'objet des critiques les plus unanimes de la part des professionnels de santé comme des patients et des institutions de la santé est celle des extensions d'indication pour les produits bénéficiant ou ayant bénéficié d'une ATU - qui constitue, selon une expression familière, le « trou dans la raquette » majeur du dispositif.
a) Un cadre juridique inadapté aux nouveaux enjeux liés au mode d'action des innovations oncologiques
(1) Les ATU sont délivrées par produit et non par indication
Les ATU sont délivrées pour un médicament innovant - en tant qu'il constitue une entité moléculaire nouvelle -, et non pour une ou plusieurs indications de ce médicament - en tant qu'il constitue un mode d'action thérapeutique.
Elles ne sont par ailleurs délivrées qu'en amont de la première AMM pour le produit considéré. Durant cette période, selon les éléments transmis par les services ministériels, « lorsqu'un médicament bénéficie d'une ATU de cohorte dans une ou plusieurs indications thérapeutiques, le directeur général de l'ANSM peut modifier l'ATU, à la demande du laboratoire, pour l'élargir à une nouvelle indication ».
Passée la délivrance de l'AMM, le périmètre de l'ATU se fige et se trouve limité au produit employé dans les indications ayant fait ou faisant l'objet de la demande d'AMM . Toute extension d'indication devient en revanche impossible au-delà de la délivrance de la première AMM.
Les premières indications demandées par le laboratoire au stade de la demande d'AMM pour leur produit deviennent de ce fait les seules autorisées jusqu'à la sortie du régime dérogatoire d'ATU.
Aux termes employés par l'INCa, dès lors que la première AMM faisant suite à un régime d'ATU de cohorte a été attribuée, seuls deux types de patients peuvent recevoir le médicament concerné en post-ATU : les patients répondant strictement aux indications de l'AMM (parfois plus étroites que celles de l'ATU de cohorte antérieure), et ceux relevant d'extensions d'indications demandées par l'industriel et en cours d'évaluation par l'agence du médicament européenne.
(2) Ce régime rencontre aujourd'hui ses limites scientifiques, entraînant d'inacceptables pertes de chances pour les patients
Les progrès scientifique, notamment dans le champ de l'oncologie, viennent questionner la pertinence de ce cadre.
L'un des domaines majeurs de développement de nouvelles molécules pour le traitement du cancer est celui de l'immunothérapie. Or, le mode d'action de ces nouveaux produits est radicalement différent des générations précédentes de médicaments anticancéreux. Dans la mesure où ils visent à renforcer le système immunitaire du patient en agissant sur des récepteurs présents dans différents organes, et non à cibler les cellules cancéreuses, ils peuvent être efficaces, de manière transversale, contre plusieurs types de cancers différents , là où les chimiothérapies actuelles visent généralement un organe particulier.
Ce mode d'action a notamment permis le développement rapide d'indications parallèles ou successives pour les anticorps anti-PD1 et anti-PDL1 . Les premières AMM pour ces produits, délivrées depuis 2011 aux États-Unis et depuis 2013 en Europe, ont ainsi successivement concerné le mélanome de stade IV, le cancer du poumon, les lymphomes de Hodgkin, les cancers de la vessie, du rein, de la tête et du cou. Le Livre blanc du Crio précité indique que des programmes de développement clinique sont en cours sur plus de 30 indications de cancers , ce qui annonce de nouvelles AMM pour les mois et les années à venir.
• Les personnes entendues par vos rapporteurs ont unanimement considéré que l'impossibilité de mettre en oeuvre une extension d'indication pour les immunothérapies sous ATU est constitutive d'importantes pertes de chances pour les patients. Le régime actuel ne garantit pas en effet aux patients de recevoir le traitement le plus efficace disponible. Des situations de besoin thérapeutique restent ainsi non couvertes.
La situation est d'autant plus absurde que les molécules concernées sont déjà présentes dans les pharmacies hospitalières, et pourraient dès aujourd'hui permettre de soigner de nombreux patients.
A notamment été cité le cas du nivolumab , qui, après avoir bénéficié d'une ATU en 2014, est aujourd'hui autorisé et admis au remboursement contre les mélanomes et les cancers du poumon. Alors même que les essais cliniques sont positifs et qu'une AMM est déjà donnée aux États-Unis dans ces indications, il est cependant impossible de le prescrire à des patients atteints, notamment, de cancers de la vessie ou ORL.
Les indications successives des nouvelles
immunothérapies anticancéreuses
et l'état des AMM et
des inscriptions au remboursement
aux États-Unis, en Europe et en
France
Source : Institut Gustave Roussy
Les patients subissent ainsi les conséquences des choix stratégiques des industriels, ou simplement de contraintes administratives et financières conduisant les laboratoires à ne pas pouvoir obtenir toutes les AMM simultanément.
Cette situation est également dommageable pour l'organisation des soins. Cette restriction est en effet susceptible de retarder l'adoption et l'appropriation par les professionnels et les établissements de santé des thérapies de demain.
L'INCa a ainsi relevé que, alors qu'elle était pionnière pour l'accès aux médicaments innovants en amont de leurs premières AMM, la France se retrouve dorénavant en « queue de peloton » pour les extensions d'indications.
Un exemple pratique illustrant cette situation a été cité à vos rapporteurs par le laboratoire MSD : si les patients français ont accédé, grâce au dispositif des ATU, au traitement d'immuno-oncologie du prembrolizumab pour le mélanome avancé 13 mois avant l'AMM européenne, ce même médicament, dans son extension d'indication pour le cancer bronchique (CBNPC), ne leur a été disponible que plus tardivement.
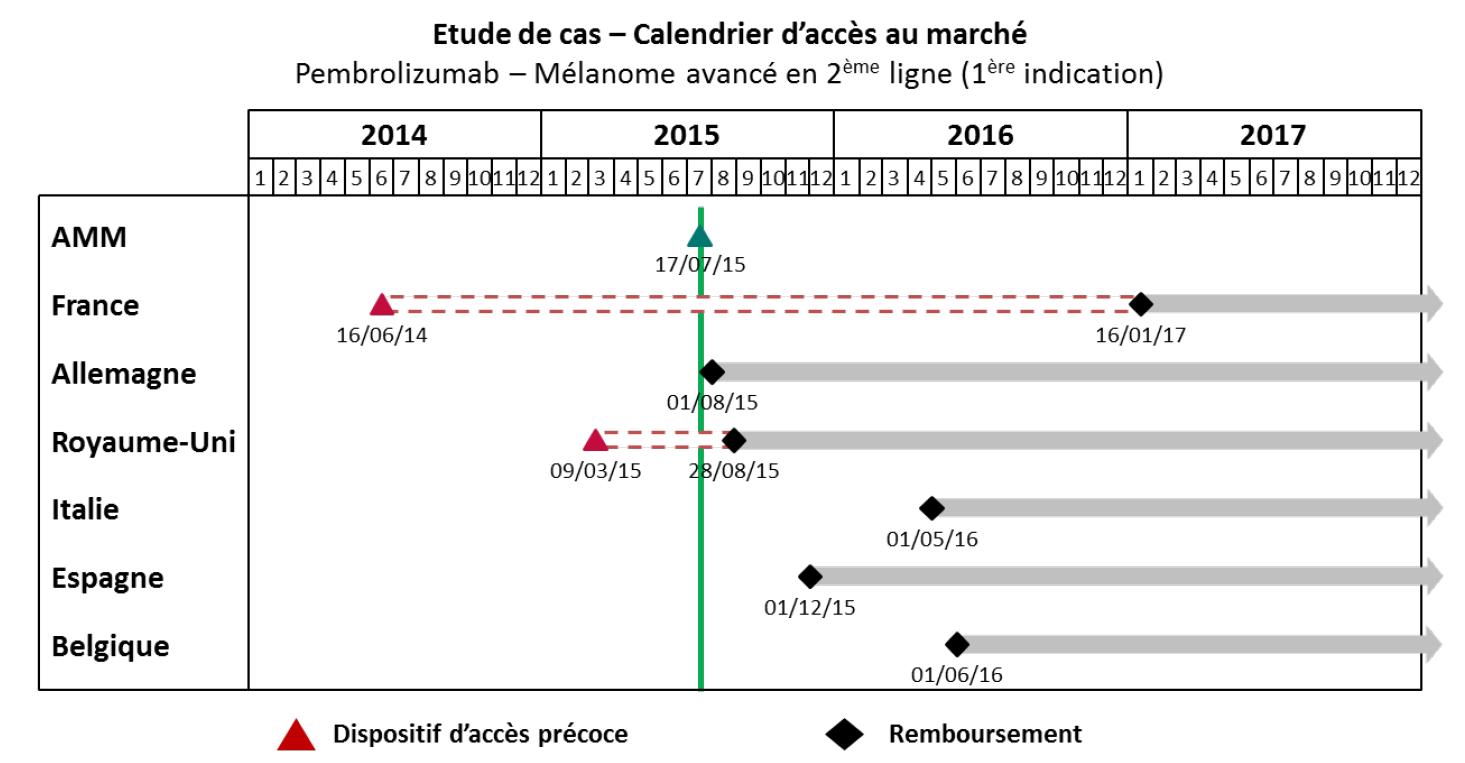
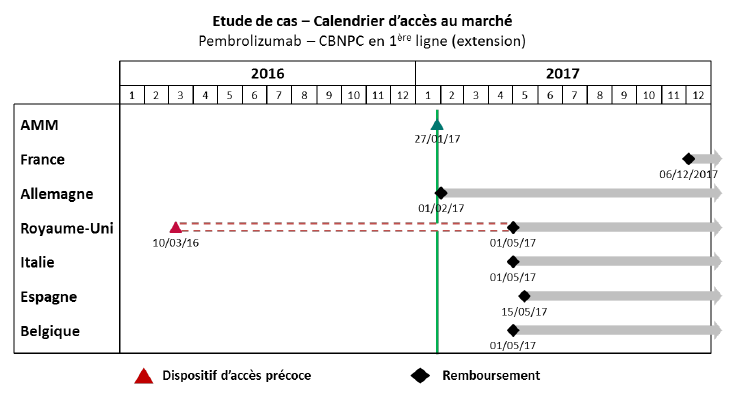
Source : MSD France
(3) Le dispositif des recommandations temporaires d'utilisation (RTU) n'apporte pas une réponse adaptée à ce besoin
• Introduites par la loi dite « Médicament » de 2011 31 ( * ) à la suite de l'affaire du Mediator®, les RTU autorisent l'utilisation d'un médicament en dehors des indications prévues par son AMM et dans le cadre d'un protocole dédié. Prévu à l'article L. 5121-12-1 du code de la santé publique, le dispositif est opérationnel depuis la parution de deux décrets d'application 32 ( * ) encadrant son régime ; il a ensuite été modifié par la LFSS pour 2014.
L'objet des RTU, accordées pour une durée de trois ans renouvelable, est d'encadrer et de sécuriser certaines pratiques de prescription constatées sur le terrain , sous condition que l'ANSM présume d'un rapport bénéfice/risque favorable dans l'indication considérée.
Une RTU ne peut être accordée qu'en l'absence d'une autre spécialité présentant le même principe actif, le même dosage et la même forme pharmaceutique, et disposant déjà d'une AMM ou d'une ATU dans l'indication considérée.
L'octroi d'une RTU emporte l'obligation pour le titulaire de l'AMM de mettre en place, à ses frais, un suivi des patients traités retraçant les données relatives à l'efficacité, à la sécurité et aux conditions d'utilisation du médicament dans l'indication considérée.
Octroyées par l'ANSM à la demande des acteurs institutionnels (autorités ministérielles, Cnam, INCa, HAS), les RTU permettent en somme d'encadrer le « bon hors AMM » . Le mécanisme vise à pallier le fait que dans de telles situations, l'industrie pharmaceutique ne demande pas toujours l'extension de l'AMM pour le produit concerné.
Dans la mesure où elles s'appliquent à des médicaments déjà autorisés et présents sur le marché, les RTU ne constituent pas à proprement parler un dispositif d'accès à des produits innovants. Dès lors cependant qu'elles pourraient permettre d'autoriser le recours à une nouvelle indication mise en évidence au cours de la vie de ces produits, elles peuvent être considérées comme un possible dispositif d'accès à l'innovation.
• Pour autant, la doctrine d'application de ce dispositif, telle qu'elle est envisagée par les différentes autorités compétentes rencontrées par vos rapporteurs, semble exclure son application au problème des extensions d'indication pour les produits ayant bénéficié d'une ATU.
Ainsi que les services ministériels l'ont rappelé, les RTU n'ont pas été conçues pour pallier cette difficulté ; du reste, en dépit de la proximité de leur dénomination, ces deux dispositifs n'ont aucun lien. Et, quand bien même on voudrait utiliser ce mécanisme à cette fin, il présenterait plusieurs faiblesses.
En premier lieu, le dispositif apparaît particulièrement lourd et rigide dans sa mise en place comme dans son fonctionnement. Il semblerait que les prescripteurs ne procèdent pas le plus souvent à l'inclusion des patients dans la RTU, du fait de contraintes administratives et procédurales jugées trop importantes.
Cette difficulté explique le faible nombre de RTU accordées depuis la mise en place du dispositif en 2012 : on en compte 47 dont 4 ont été arrêtées du fait de l'octroi d'une AMM dans l'indication considérée. Deux d'entre elles ont fait l'objet d'un traitement médiatique plus marqué : l'utilisation du Baclofène® dans le traitement de l'alcoolodépendance (mars 2014), ainsi que celle de l'Avastin® dans la dégénérescence maculaire liée à l'âge (DMLA) dans sa forme néovasculaire (juin 2015).
En outre, les RTU ne sont pas délivrées à la demande des laboratoires, ce qui exclut du circuit l'acteur majeur du développement des produits.
Enfin et surtout, un recours renforcé au mécanisme des RTU ainsi que son ouverture aux laboratoires pourraient conduire à déséquilibrer les négociations de prix conduites entre le CEPS et les industriels . La mise en oeuvre d'une RTU postérieurement à la conclusion de ces discussions aboutirait en effet à augmenter la population cible des médicaments concernés, et donc le volume de produits pris en charge par l'assurance maladie, sans que cette donnée ait été prise en compte dans la fixation préalable du prix. La demande d'une AMM dans une indication cible étroite puis d'une RTU visant une population plus large pourrait ainsi alimenter les stratégies industrielles, pour un impact financier certain sur les comptes de l'assurance maladie.
• Vos rapporteurs souscrivent cependant à l'observation des équipes hospitalières et de la HAS selon laquelle le dispositif des RTU est sous-exploité alors même qu'il pourrait répondre à d'importants besoins . Il peut par ailleurs constituer un levier d'incitation pour la mise en place d'essais cliniques par les laboratoires dans le but de parvenir à une extension d'indication de leur médicament.
Quoique cette question ne relève pas directement de l'objet du présent rapport, ils estiment dès lors indispensable de le rendre à la fois plus rapide, plus souple et plus simple , par exemple par l'introduction de délais maximum de procédure.
b) Adapter les ATU pour couvrir les besoins thérapeutiques liés aux extensions d'indication
Vos rapporteurs estiment en tout état de cause indispensable de mettre en place dans les meilleurs délais un élargissement du dispositif d'ATU aux nouvelles indications de produits disposant déjà d'une AMM et faisant l'objet de développements innovants dans de nouvelles indications , en cas de rapport bénéfices/risques satisfaisant et en l'absence d'alternative thérapeutique.
Suivant la piste avancée par la plupart des acteurs entendus, la mise en place d'un système d'ATU par indications apparaît comme la façon la plus simple et efficace de répondre à cet objectif. Cette solution reviendrait à repenser le principe du dispositif pour l'attribuer non plus une seule fois pour un produit dans quelques indications limitativement énumérées, mais, potentiellement, plusieurs fois successivement pour un même produit.
|
Proposition n° 3 : Délivrer les ATU par indication et non plus par produit, de manière à couvrir les situations d'extension d'indication survenant après la délivrance de la première AMM. |
4. Revenir sur un régime de régulation financière devenu excessivement complexe
a) Un encadrement financier renforcé dans le cadre des dernières LFSS
L'évolution du coût du dispositif des ATU a justifié un renforcement de son encadrement financier.
En particulier, l'article 97 de la loi de financement de la sécurité sociale pour 2017 33 ( * ) a été adopté dans l'objectif affiché par le précédent gouvernement de « préserver » les dispositifs d'ATU et de post-ATU en assurant leur soutenabilité financière pour l'assurance maladie. Il s'agissait de mettre en place un « accès précoce plus régulé », reposant sur « un partage des coûts entre l'industriel et la collectivité ».
Cet encadrement repose sur deux leviers.
• D'une part, un plafonnement de la dépense moyenne annuelle par patient au titre des produits sous ATU, selon un mécanisme à double détente : pour tout produit dont le chiffre d'affaires excède 30 millions d'euros par an, le coût annuel par patient est limité à 10 000 euros .
Le cas échéant, ce plafonnement se traduit par un remboursement rétroactif annuel à l'assurance maladie, par le laboratoire pharmaceutique, de la part de son chiffre d'affaires en dépassement de ces seuils, sous la forme d'une remise.
Selon les services ministériels, il s'agit d'un « garde-fou » visant à mettre fin au régime de totale liberté dont bénéficiaient les industriels dans la fixation de l'indemnité au titre des produits sous ATU , dans la mesure où il affaiblissait ensuite le pouvoir de négociation du CEPS dans la fixation du prix, en partant d'un niveau de référence élevé.
Applicable aux produits pour lesquels une ATU a été octroyée à compter du 1 er janvier 2017, ce mécanisme n'a, selon les services ministériels, encore jamais trouvé à s'appliquer.
Vos rapporteurs observent que l'absence de dépassement à ce jour de ce plafond signifie que les industriels ont probablement calibré le montant de l'indemnité demandée afin de l'inscrire dans la fourchette fixée par la loi, ce qui constitue une ébauche de régulation. Ils relèvent également que, selon les indications fournies par l'assurance maladie, ce système de plafonnement du coût des traitements onéreux existe dans la plupart des pays comparables à la France.
• D'autre part, un nouveau mode de calcul pour le montant du remboursement rétroactif du différentiel entre l'indemnité ATU fixée par l'industriel et le prix du médicament défini par le CEPS après son AMM.
L'article 97 a modifié le mode de calcul du montant rétroactivement reversé par le laboratoire à l'assurance maladie, sous forme de remise, dans tous les cas où le prix fixé par le CEPS est inférieur à l'indemnité déterminée par le laboratoire en ATU et post-ATU.
Ce montant est désormais calculé non plus par rapport au prix ou au tarif de remboursement (le « prix facial »), mais par rapport au « prix net de référence » (prix net des remises conventionnellement consenties par le laboratoire) pour le produit concerné.
Tableau comparatif des dispositions de l'article
L. 162-16-5-1
avant et après la réforme introduite par
l'article 97 de la LFSS pour 2017
|
[...] le laboratoire reverse aux organismes mentionnés à l'article l. 213-1 désignés par le directeur de l'agence centrale des organismes de sécurité sociale, sous forme de remise, |
|
|
avant la LFSS pour 2017 la différence entre le chiffre d'affaires facturé aux établissements de santé sur la base de l'indemnité et celui qui aurait résulté de la valorisation des unités vendues au prix ou au tarif de remboursement fixé par le comité . |
après la LFSS pour 2017 la différence entre le chiffre d'affaires facturé aux établissements de santé, au titre de la période s'étendant de l'obtention de l'autorisation mentionnée à l'article l. 5121-12 du code de la santé publique à la première date d'inscription au remboursement, minoré le cas échéant des remises mentionnées au ii du présent article au titre de cette même période, et celui qui aurait résulté de la valorisation des unités vendues au prix net de référence . |
Aux termes de l'article L. 162-18 du code de la sécurité sociale, ce prix net de référence est calculé à partir du niveau prévisionnel des prix nets de remises sur les trois années à venir . Il repose donc sur une prévision des volumes de vente pour les trois années suivant la sortie du dispositif ATU . Selon les indications fournies par le CEPS, il s'agit en somme de défalquer du prix facial le montant des remises conventionnelles qui pourraient être dues au titre de ces trois années - étant précisé qu'un prix net plus bas que celui qui résulterait de ce mode de calcul peut toujours être déterminé par voie conventionnelle.
Ce mécanisme, applicable aux chiffres d'affaires réalisés dès l'année 2016, viserait avant tout à inciter les laboratoires à conclure rapidement un accord de prix permettant la sortie du post-ATU .
La Cnam a qualifié le nouveau mode de calcul de « nécessité absolue » du fait de l'impact financier du régime des ATU. Selon la DSS, ce mécanisme se justifie par les prétentions très élevées des laboratoires en matière de prix . Ce mode de calcul a dès lors été conçu, comme le mécanisme de plafonnement, pour inciter les industriels à sortir rapidement du régime financier dérogatoire de l'ATU et du post-ATU.
Il ne semble pas, pour autant, que cet objectif soit atteint dans la mesure où il paraît contribuer, à l'opposé, à une crispation des positions.
b) Des dispositifs illisibles et contestés : des modalités à simplifier
Vos rapporteurs entendent les raisons qui ont justifié le renforcement de la régulation financière du dispositif des ATU, dans un contexte de croissance dynamique des coûts et de l'arrivée de nouvelles innovations dont les prix, réels ou fantasmés, sont très élevés.
Pour autant, les modalités prévues par l'article 97 de la LFSS pour 2017 soulèvent plusieurs difficultés.
• Le mécanisme de plafonnement aboutit, pour les industriels, à une dégradation de la valeur marchande de l'innovation .
On peut s'interroger en effet sur l'adaptation du seuil de 10 000 euros par patient à certains traitements innovants : les représentants d'Unicancer ont estimé qu'il n'était « absolument pas adapté aux nouvelles thérapies anticancéreuses », et donc susceptible de fragiliser le dispositif des ATU.
• Mais c'est avant tout le mode de calcul de l'indemnité rétroactive versée sous forme de remise par les laboratoires au sortir de la phase de post-ATU qui concentre les critiques.
Qualifié par les représentants du Leem de « mécanisme surréaliste », ce nouveau régime a, de l'avis unanime des industriels entendus par vos rapporteurs, fortement dégradé l'attractivité des ATU .
Outre que cette modification, prévue par voie d'amendement à un stade avancé de la procédure d'adoption de la LFSS pour 2017, pose la question générale de la sécurité juridique du système des ATU , la complexité de ce nouveau mode de calcul apparaît très dissuasive pour les laboratoires. Cette complexité est d'autant plus mal reçue qu'elle se justifie par des raisons strictement économiques, voire comptables - ce dont les autorités ministérielles ne se cachent d'ailleurs pas -, perçues comme non justifiées au regard des enjeux de promotion de l'innovation.
Surtout, ce mode de calcul est dénoncé comme remettant en cause la lisibilité et la prévisibilité du mécanisme de sortie des ATU , dans la mesure où le prix net qui sert de base à la détermination de la remise est calculé sur la base de volumes prévisionnels de vente . Les laboratoires estiment que cela revient à mettre en place un mécanisme de remise sur un chiffre d'affaires potentiel , qui est par nature incertain et soumis à de nombreux aléas - comme par exemple l'arrivée sur le marché d'un nouveau traitement concurrent.
Les industriels soulignent par ailleurs que le montant de la remise ainsi calculée est très difficile à absorber par les petits laboratoires de biotechnologies , qui constituent aujourd'hui des acteurs importants de l'innovation médicamenteuse, et d'autant plus lorsqu'ils sont monoproduit.
Ces difficultés sont renforcées par le caractère rétroactif du dispositif , qui, voté en 2017, s'applique aux chiffres d'affaires réalisés au titre des ATU en 2016. Les entreprises concernées n'ont dès lors pas toujours pu provisionner les sommes à reverser.
Les industriels avancent enfin que le raffinement technique de nouveau dispositif est très difficile à expliquer auprès des instances décisionnaires de celles des entreprises concernées dont la taille est mondiale et dont le siège se situe à l'étranger.
Vos rapporteurs notent que ces difficultés s'ajoutent à celles résultant du mode de calcul de la contribution sur le chiffre d'affaires des laboratoires , dite « clause de sauvegarde » ou « L » et prévue par l'article L. 138-10 du code de la sécurité sociale. Le montant de cette taxe, qui porte sur la progression du chiffre d'affaires des laboratoires d'une année sur l'autre, est en effet calculé sur la base d'un chiffre d'affaires net de remises pour l'année n-1, ce qui conduit à un gonflement artificiel du chiffre d'affaires pour l'année n et à une augmentation mécanique du montant de la contribution. Cette situation a été dénoncée au cours de l'examen des derniers PLFSS par votre commission des affaires sociales, qui a adopté à plusieurs reprises un amendement tendant à rendre comparables les assiettes prises en compte d'une année sur l'autre pour le calcul de la contribution due au titre de la clause de sauvegarde.
Cette contribution a été scindée, dans le cadre de la LFSS pour 2017, en une contribution Lh portant sur la progression du chiffre d'affaires réalisé à l'hôpital et une contribution Lv pour les médicaments de ville. Or, la plupart des médicaments innovants étant distribués à l'hôpital par un petit nombre de laboratoires, le montant de la contribution à leur charge s'en trouve mécaniquement augmenté.
• Vos rapporteurs estiment que, du fait de la particulière complexité du dispositif mis en place par l'article 97 de la LFSS pour 2017, il n'apparaît pas raisonnable de poursuivre son application .
L'objectif de maîtrise des dépenses de l'assurance maladie doit être concilié avec la nécessaire attractivité du dispositif d'ATU, afin de continuer à garantir aux patients cet accès essentiel à l'innovation médicamenteuse.
Compte tenu de l'allongement de la phase de post-ATU, qui expose l'assurance maladie au paiement d'une indemnité très élevées pendant une longue période, vos rapporteurs considèrent cependant indispensable de réfléchir à un nouveau mécanisme de régulation .
La proposition n° 1 formulée dans le cadre du présent rapport apporte un élément de réponse en garantissant l'adéquation du niveau de l'indemnité payée par l'assurance maladie en ATU avec les résultats thérapeutiques réellement observés. Des professionnels de santé entendus ont d'ailleurs souligné que, en toute logique, l'indemnité versée au titre des ATU devrait d'abord être faible, avant d'être révisée à la hausse en fonction des résultats obtenus.
|
Proposition n° 4 : Revenir sur le mode de calcul complexe de la remise rétroactivement versée par les laboratoires au titre de la récupération sur l'indemnité de la phase d'ATU, tel que prévu par l'article 97 de la LFSS pour 2017. |
*
Les difficultés relatives à la mise en oeuvre des ATU ne se comprennent que dans le cadre plus général des procédures de droit commun de mise sur le marché et de fixation des prix .
Les préconisations faites par les industriels traduisent au fond la volonté d'une valorisation commerciale rapide des innovations, aujourd'hui rendue difficile par l'allongement de la phase de post-ATU.
C'est finalement au moins autant le cadre général de l'évaluation des médicaments et de la fixation des prix, dont les délais apparaissent incompatibles avec l'accélération des innovations scientifiques, qui doit aujourd'hui évoluer.
II. FLUIDIFIER L'ACCÈS AUX MÉDICAMENTS INNOVANTS APRÈS LEUR AUTORISATION DE MISE SUR LE MARCHÉ
Si le dispositif des ATU constitue, quelles qu'en soient les adaptations nécessaires, une force indéniable du modèle français pour l'accès précoce des patients aux innovations, dans les autres cas la situation est perçue comme moins satisfaisante.
Dans un contexte d'innovation dynamique, un grand nombre d'intervenants ont pointé les lenteurs et lourdeurs affectant l'accès « de droit commun » au marché français des médicaments innovants :
- d'une part, les délais entre l'autorisation de mise sur le marché et la mise à disposition effective aux patients sont plus longs que dans d'autres pays, même si ces inquiétudes sont à relativiser du fait des autres canaux d'accès précoce aux innovations ( via les ATU ou les essais cliniques) ;
- d'autre part, les procédures administratives séquencées précédant cette mise à disposition, indispensables pour définir les conditions de prise en charge solidaire des traitements innovants, soulèvent des interrogations, parfois sources d'incompréhensions entre les acteurs voire révélatrices d'un rapport de force autour des enjeux de régulation des prix.
Il ressort, plus globalement, le sentiment d'un système de santé qui subit l'innovation , et son coût, plutôt que de l'anticiper et de l'accompagner, en repensant l'équilibre délicat entre l'ambition d'un accès de plus en plus précoce à certains innovations prometteuses et les exigences tout aussi essentielles de sécurité des patients et de maîtrise budgétaire.
Si des évolutions se dessinent déjà, des mesures de fond seraient à envisager pour favoriser l' « accueil » des innovations, en renforçant la lisibilité ou la souplesse des procédures et la visibilité pour les acteurs.
A. AU-DELÀ DES LOURDEURS RÉGLEMENTAIRES, UN CLIMAT DE DÉFIANCE ET D'INCERTITUDES
1. L'accès au marché de droit commun des médicaments : une procédure séquencée et à forts enjeux
a) L'autorisation de mise sur le marché : un examen centralisé au niveau européen pour les médicaments innovants
En dehors de la procédure d'ATU ou de dispositifs d' early access équivalents, la commercialisation des médicaments et leur mise à disposition auprès des patients ont comme point de départ l'autorisation de mise sur le marché (AMM), accordée après évaluation de l'efficacité, de la sécurité et du bénéfice/risque dans une indication donnée ; elle est requise pour les « médicaments à usage humain destinés à être mis sur le marché dans les États membres et préparés industriellement ou fabriqués selon une méthode dans laquelle intervient un processus industriel. » 34 ( * )
Pour les médicaments présumés innovants, la procédure est centralisée au niveau de l'agence européenne du médicament ( European Medicines Agency - EMA) : l'AMM délivrée par la commission européenne après avis de l'EMA est unique pour l'ensemble du territoire de l'Union européenne.
|
Les procédures d'autorisation de mise sur le marché des médicaments en Europe Il existe quatre procédures d'autorisation de mise sur le marché des médicaments : une procédure nationale et trois procédures européennes. • La procédure centralisée au niveau européen, qui relève de l'EMA, est obligatoire pour les médicaments de thérapie innovante, ceux issus des biotechnologies, les médicaments contenant une nouvelle substance active et dont l'indication thérapeutique est le traitement de certaines affections (SIDA, cancer, maladie neurodégénérative, diabète, maladies auto-immunes et maladies virales) ainsi que les médicaments orphelins indiqués dans le traitement des maladies rares. Pour les autres pathologies, elle reste optionnelle. Cette procédure peut également être envisagée si le médicament présente un intérêt majeur pour les patients. En 2016, 114 AMM ont été délivrées en procédure centralisée par l'EMA dont 14 concernant des médicaments orphelins. L'agence française, l'ANSM, a été rapporteur ou co-rapporteur de 14 dossiers sur 114 (soit 12 %). • La procédure décentralisée s'applique pour les médicaments qui ne sont pas encore autorisés dans l'Union européenne et sont destinés à au moins deux États membres. L'industriel demande à un État membre d'agir en tant qu'état de référence parmi les États dans lesquels il souhaite commercialiser son médicament. • La procédure de reconnaissance mutuelle est fondée sur la reconnaissance d'une AMM déjà accordée dans un des États membres de l'Union européenne, appelé « État de référence », par d'autres États membres désignés par le laboratoire pharmaceutique titulaire de l'AMM. Dans ces deux cas (procédure décentralisée et de reconnaissance mutuelle), ce sont les autorités nationales compétentes (en l'occurrence l'ANSM en France) qui délivrent les AMM dont les annexes sont harmonisées. • La procédure nationale concerne des médicaments autorisés uniquement dans le pays concerné, ce qui est notamment le cas de génériques. En 2016, le nombre d'AMM délivrées par l'ANSM s'établit à 565 au total (502 en 2015) dont 245 AMM en procédure nationale, 295 en procédure décentralisée et 25 en procédure de reconnaissance mutuelle ; 72 % (soit 406 AMM) concernant des médicaments génériques. Source : ANSM - rapport d'activité 2016 (20 septembre 2017) |
Comme cela a été relevé à vos rapporteurs, les États-Unis sont aujourd'hui le premier marché pour la commercialisation d'un grand nombre de médicaments innovants 35 ( * ) . Toutefois, les enjeux de l'accès précoce ont conduit l'EMA, à l'image de son homologue américaine (la Food and Drug Administration - FDA), à mettre en place des outils pour accélérer le traitement des demandes d'AMM relevant de la procédure centralisée et plus généralement pour encourager l'accès précoce.
|
L'accès précoce aux médicaments en Europe Plusieurs outils ont été mis en place au niveau de l'Agence européenne du médicament pour favoriser l'accès précoce des patients aux nouveaux traitements éligibles à la procédure centralisée : - l'évaluation accélérée, qui peut être sollicitée par l'industriel pour des médicaments présentant un intérêt majeur du point de vue de la santé publique : cette procédure permet de réduire le délai maximum à 150 jours au lieu de 210 ; - l'octroi d'AMM conditionnelle ; cette procédure peut concerner des médicaments pour le traitement de maladies graves ou des situations d'urgence en réponse à des menaces sur la santé publique ou pour des médicaments dits orphelins ; elle requiert des données moins complètes que celles exigées dans la procédure de droit commun, en contrepartie de certaines obligations ; - l'usage compassionnel (programmes d' early access ) permettant aux États membres de mettre en place des dispositifs tels que les ATU en France ; - le programme PRIME ( Priority Medicines ) de soutien au développement de médicaments visant un besoin médical non satisfait ; - le programme Adaptative pathways issu d'un projet pilote de 2014, permettant l'autorisation précoce pour une population très réduite et une indication étroite, suivie de nouvelles phases itératives de développement cliniques en vue d'élargir progressivement l'indication (AMM dites « fractionnées »). |
Ces évolutions traduisent une volonté de gagner en réactivité dans un contexte dynamique d'innovation et donc une large prise de conscience des enjeux de l'accès précoce. Elles ne vont pas toutefois sans soulever des critiques et réinterrogent les procédures « classiques » d'évaluation qui suivent au niveau national pour déterminer l'accès au remboursement et le prix, pays par pays.
b) Un cadre national rigoureux en vue de la prise en charge des médicaments par la solidarité nationale
(1) La phase d'évaluation par la Haute Autorité de Santé (HAS) : une procédure lourde mais essentielle
L'évaluation scientifique précédant l'AMM est doublée, lorsque le laboratoire qui commercialise un médicament souhaite obtenir son inscription sur la liste des produits remboursables, d'une autre procédure d'évaluation thérapeutique et, dans certains cas, médico-économique. Elle est assurée en France par une autorité administrative indépendante, la Haute Autorité de santé (HAS).
• L'évaluation thérapeutique des médicaments relève plus spécifiquement de la commission de la transparence de la HAS, instance scientifique indépendante composée de vingt-et-un experts avec voix délibérative (médecins, pharmaciens, cliniciens, spécialistes en méthodologie et épidémiologie, membres des associations de patients et d'usagers) 36 ( * ) .
Cette commission est chargée de donner un avis aux ministres chargés de la santé et de la sécurité sociale en vue de la prise en charge des médicaments et de la fixation de leur prix , en appréciant :
- d'une part, le service médical rendu (SMR) qui sert à déterminer si un médicament doit être remboursé et à quel taux ;
- d'autre part, l' amélioration du service médical rendu (ASMR) pris en compte pour la fixation du prix.
|
SMR, ASMR : de quoi parle-t-on ? • Le service médical rendu (SMR) : Le médicament a-t-il suffisamment d'intérêt clinique pour être pris en charge par la solidarité nationale ? L'appréciation de ce critère prend en compte plusieurs aspects : - la gravité de la pathologie pour laquelle le médicament est indiqué, - l'efficacité et les effets indésirables du médicament, - sa place dans la stratégie thérapeutique notamment au regard des autres thérapies disponibles, - le caractère préventif, curatif ou symptomatique du médicament, - son intérêt pour la santé publique (gravité de la maladie et prévalence, besoin médical, impact sur la qualité de vie, impact sur l'organisation des soins...). Il existe plusieurs niveaux de SMR qui apportent un éclairage scientifique et clinique à l'Uncam et au ministre en charge de la santé pour déterminer si un médicament doit être remboursé et à quel taux : - SMR majeur ou important taux de remboursement 65 % - SMR modéré taux de remboursement 30 % - SMR faible taux de remboursement 15 % - SMR insuffisant pour justifier une prise en charge par la collectivité. Le SMR d'un médicament est mesuré à un moment donné. Il peut évoluer dans le temps et son évaluation se modifier. • L'amélioration du service médical rendu (ASMR) : Le médicament apporte-t-il un progrès par rapport aux traitements disponibles ? L'ASMR correspond au progrès thérapeutique que le médicament est susceptible d'apporter par rapport aux traitements déjà disponibles, au regard : - des données comparatives disponibles en termes d'efficacité et de tolérance (niveau de preuve, quantité d'effet, extrapolation en pratique clinique), - du besoin thérapeutique et de sa couverture, - de l'impact sur la qualité de vie. Les différents niveaux d'ASMR apportent un éclairage scientifique et clinique au CEPS et au ministre pour la fixation du prix des médicaments : - ASMR I, majeure, - ASMR II, importante, - ASMR III, modérée, - ASMR IV, mineure, - ASMR V, inexistante, signifie « absence de progrès thérapeutique » : dans ce cas, le médicament ne peut être inscrit au remboursement que s'il apporte une économie dans les coûts de traitement. Source : HAS |
La commission de la transparence de la HAS évalue (en première inscription ou pour des extensions d'indication) ou réévalue près de 800 produits chaque année. Entre 2014 et 2017, elle a eu à évaluer 261 nouveaux médicaments en première indication.
• L'évaluation médicale peut être complétée, dans certains cas encore rares, d'une évaluation médico-économique , qui met en regard le coût du médicament et ses conséquences sur l'organisation des soins.
Cette mission relève depuis 2008 de la commission d'évaluation économique et de santé publique (CEESP) de la HAS, dont la composition est différente de celle de la commission de la transparence 37 ( * ) . Celle-ci formule un avis sur le rapport coût/efficacité des actes, médicaments ou dispositifs médicaux susceptibles d'avoir un impact significatif sur les dépenses de l'assurance maladie. Elle est prise en compte comme critère de fixation du prix du médicament depuis 2012.
Pour les médicaments, cette procédure s'applique à ceux réputés les plus innovants (ASMR I à III) et pouvant présenter un impact « significatif » sur les dépenses de l'assurance maladie, évalué à plus de 20 millions d'euros les deux premières années de commercialisation.
Le nombre de produits concernés reste en pratique limité : vingt avis d'efficience de produits de santé ont été rendus en 2016 et autant en 2017 38 ( * ) .
(2) La phase de fixation du prix : une démarche conventionnelle qui s'inscrit dans un cadre budgétaire contraint
Sur la base des résultats de l'évaluation et notamment de l'ASMR, s'engage ensuite la négociation ou la fixation du prix du médicament devant le comité économique des produits de santé (CEPS) .
Le CEPS est un organisme interministériel placé sous l'autorité conjointe des ministres chargés de la santé, de la sécurité sociale et de l'économie. Sa formation délibérante en matière de médicament est composée, outre son président et son vice-président, de cinq représentants de l'administration 39 ( * ) , de trois représentants de l'assurance maladie obligatoire et d'un représentant des complémentaires.
Le mode de fixation des prix se fonde sur les dispositions prévues par le code de la sécurité sociale, mais également sur un accord-cadre signé le 31 décembre 2015 entre le CEPS et le Leem pour trois ans, une lettre d'orientation ministérielle en date du 17 août 2016 et une « doctrine » développée par le CEPS et formalisée dans son rapport annuel d'activité.
Le prix du médicament est fixé par voie de conventions bilatérales entre le CEPS et les entreprises pharmaceutiques.
Le présent rapport n'a pas vocation à examiner en détail ce processus complexe, qui a déjà fait l'objet d'un rapport de votre commission des affaires sociales publié en juin 2016 40 ( * ) et dont la Cour des comptes s'est encore récemment saisie 41 ( * ) .
Il est toutefois utile d'en rappeler quelques principes généraux.
|
La fixation du prix du médicament : principes généraux • Des dispositions distinctes du code de la sécurité sociale s'appliquent aux modalités de fixation de prix des médicaments, selon que le celui-ci est dispensé en officine ou à l'hôpital, et selon les différentes modalités de prise en charge (liste en sus 42 ( * ) , médicaments rétrocédables). Selon les articles L. 162-16-4 (médicaments vendus en officine) et L. 162-16-6 (liste en sus) du code de la sécurité sociale, la fixation du prix « tient compte principalement de l'amélioration du service médical rendu par le médicament, le cas échéant des résultats de l'évaluation médico-économique , des prix des médicaments à même visée thérapeutique , des volumes de vente prévus ou constatés ainsi que des conditions prévisibles et réelles d'utilisation du médicament. » • L'économie générale de la fixation des prix est articulée autour de « prix faciaux » sur lesquels s'appliquent des « remises conventionnelles » qui abaissent le « prix net » des médicaments. Le prix net n'est pas public, le montant des remises étant couvert par le secret des affaires. Les remises conventionnelles (article L. 162-18 du code de la sécurité sociale) sont négociées entre le CEPS et les entreprises pharmaceutiques et peuvent être conclues pour tous les médicaments quelle que soit leur ASMR. Leur montant brut, sur la base des ventes réalisées en 2016, s'établit d'après le CEPS à 1 milliard d'euros. 44 % de ces remises portent sur une dizaine de produits. Les engagements de type prix/volume représentent 41 % des remises dues. Ces remises sont recouvrées par les Urssaf pour le compte de l'assurance maladie. Celles liées à plusieurs catégories de médicaments pris en charge par l'assurance maladie (liste en sus, médicament sous et post-ATU, rétrocessions hospitalières) viennent alimenter en recette le fonds de financement de l'innovation pharmaceutique (FFIP) créé par l'article 95 de la loi de financement de la sécurité sociale pour 2017. Géré par la Cnam, ce fonds vise à lisser les dépenses de médicaments innovants et coûteux mais conduit à sortir ces dépenses du périmètre de l'Ondam. • Pour les médicaments innovants, une garantie de prix européen est prévue depuis 2003. Elle consiste à accorder à certains médicaments, pour une durée de cinq ans, un prix hors taxe qui ne peut être inférieur au plus bas prix pratiqué par un panel de quatre pays européens (Allemagne, Espagne, Italie, Royaume-Uni). Cette garantie ne concerne que le prix facial . Celle-ci s'applique, sauf exception justifiée, à l'ensemble des médicaments avec une ASMR I à III et, sous certaines conditions, à des produits avec une ASMR IV. Pour être éligible à cette garantie de prix le médicament doit avoir recueilli un avis de la commission d'évaluation économique de la HAS permettant au CEPS de recueillir les conditions de son efficience médico-économique. • Pour les médicaments hospitaliers , achetés par les hôpitaux, ceux de la liste en sus ont un prix (tarif de responsabilité) fixé par convention avec le CEPS (tarif déclaré par l'entreprise au CEPS qui a la possibilité de s'y opposer et de fixer son propre tarif) mais laissant la possibilité aux établissements de négocier un prix inférieur. Pour les spécialités rétrocédées, le prix de vente aux établissements de santé est déclaré par l'entreprise au CEPS qui peut s'y opposer et fixer ce prix. Sources : Rapport d'activité du CEPS pour 2016 et Cour des comptes (Ralfss 2017) |
(3) Des choix d'organisation différents entre pays
Ces étapes successives en vue de la commercialisation des médicaments et de leur admission au remboursement - AMM, évaluation, fixation du prix - visent à garantir la pleine sécurité des patients et à déterminer les conditions de leur prise en charge par la solidarité nationale.
Toutefois, comme l'ont relevé les représentants du Leem, cette procédure est comparable, en France, à « une succession de haies ».
D'autre pays ont opté pour un processus plus fluide d'accès au marché. C'est notamment le cas de l' Allemagne , fréquemment cité en exemple à vos rapporteurs : les médicaments sont disponibles dès l'obtention de l'AMM , sur la base d'un prix librement fixé par les industriels. La procédure d'évaluation en vue de la détermination du prix et de l'admission au remboursement intervient dans un délai d'un an suivant cette mise sur le marché et se déroule parallèlement à celle-ci.
Si ce modèle apparaît attractif, il faut noter qu'il fait peser une pression forte sur la procédure d'évaluation et de fixation du prix. Son attractivité au regard du modèle français est à tempérer en outre par l'existence en France des ATU.
D'une manière générale, si les modèles étrangers sont évidemment utiles à la réflexion, il est pour vos rapporteurs délicat voire non pertinent d'envisager toute transposition, tant les organisations des systèmes de santé et leur mode de régulation diffèrent par ailleurs.
|
Les procédures d'accès au marché des médicaments innovants en Europe : l'exemple de l'Allemagne, du Royaume-Uni et de l'Italie Allemagne La procédure de fixation du prix des médicaments innovants a été réformée par une loi entrée en vigueur en 2011. Le prix est fixé librement par le laboratoire pour la seule première année de mise sur le marché suivant l'AMM . Au-delà, le prix est négocié avec l'assurance maladie, sur la base de l'efficacité. Le processus dure douze mois : - dans les trois mois suivant l'accès au marché, l'institut pour la qualité et l'efficience des services de santé (IQWiG), organisme scientifique indépendant, doit avoir procédé à l'évaluation du médicament ; - dans les six mois suivant l'accès au marché, le Comité fédéral conjoint (G-BA), collège associant les fédérations de médecins, d'hôpitaux et des caisses d'assurance maladie, statue sur l'amélioration du service médical rendu : - si le médicament n'apporte pas de valeur ajoutée, l'assurance maladie peut fixer le montant remboursé de ce médicament en fonction d'un groupe de médicaments comparable. Les patients doivent payer l'éventuelle différence entre ce prix de référence et le prix de vente lorsque ce dernier est supérieur ; - si le médicament démontre une valeur ajoutée clinique, son niveau de remboursement fait l'objet de négociations entre le GKV-SV (représentant des fonds d'assurance maladie au niveau national) et les laboratoires pharmaceutiques, en liaison avec les représentants des assurances privées. L'Allemagne privilégie la négociation d'un montant maximal de remboursement, basée sur les prix européens, le prix des médicaments comparables, la valeur ajoutée et la population cible. Si aucun accord n'est trouvé à l'issu du délai de 12 mois à compter de la mise sur le marché, une commission d'arbitrage fixe le montant de remboursement, dans un délai de maximum trois mois, qui s'appliquera rétroactivement dès le treizième mois après la mise sur le marché du produit. Dès l'AMM, les hôpitaux peuvent acheter et utiliser les médicaments, pris en charge sur la base d'un montant forfaitaire, avec des mécanismes de financements additionnels pour des médicaments particulièrement onéreux. • Royaume-Uni Les prix sont fixés librement par les laboratoires mais une autre forme de régulation existe. Le National Institute for Health and Care Excellence (NICE), organisme indépendant, ne procède pas à l'évaluation de tous les médicaments ; l'évaluation est établie à l'initiative du Department of Health , dans un délai de 20 semaines. Elle se fonde notamment sur le rapport coût-efficacité du médicament (sur la base de l' Incremental Cost Effectiveness Ratio ou ICER, qui doit être situé entre 20 000 et 30 000 £) ; elle conduit à une recommandation de prise en charge par le NHS, le cas échéant avec restrictions, ou un refus , le cas échéant temporaire, de prise en charge (dans ce dernier cas le NHS n'a aucune obligation de mettre à disposition le produit pour les patients). Afin de pallier cette difficulté, le Cancer Drug Fund créé en 2011 permet de financer des médicaments anticancéreux qui n'ont pas été recommandés par le NICE. Ce dispositif a rencontré des limites en termes de financement, avec un budget initial de 200 millions de livres passé à 340 millions de livres en 2015 ; de nombreuses molécules et/ou indications ont été radiées. Le dispositif a été réformé en 2016 et réintégré dans le processus d'évaluation par le NICE, qui peut recommander le produit pour son utilisation via le Cancer drugs fund . L'évaluation initiale se fait de manière anticipée, avant l'AMM, avec un financement et un accès temporaires pour les patients dans l'attente de l'évaluation finale. • Italie Pour les médicaments classés comme remboursables par l' Agenzia Italiana del Farmaco (AIFA), la fixation du prix est déterminée, depuis 2004, après négociation avec le laboratoire. L'évaluation du taux de remboursement et du prix est réalisée sur la base du ratio coût-efficacité du produit, du bénéfice-risque, de l'impact financier prévisible, des volumes prévisionnels de vente, des prix et des consommations dans les autres pays européens. Les prix sont habituellement fixés pour deux ans. Si aucun accord sur le prix n'est trouvé, le médicament est reclassifié en non-remboursable. Les médicaments décrits comme des innovations thérapeutiques sont inscrits automatiquement sur les formulaires régionaux (cette étape supplémentaire est liée au co-paiement assuré par les autorités régionales). Depuis 2011, l'AIFA a fixé un prix de remboursement maximum pour les médicaments remboursables (classe A). Ce « cap » est basé sur une enquête des prix en cours dans les pays de l'Union Européenne. Le recours à des contrats de performance ou de partage de risques est particulièrement développé pour les médicaments onéreux, avec un accent sur le suivi en vie réelle via des registres nationaux. D'après l`INCa, en 2015 environ 200 millions d'euros soit 1 % des dépenses de médicaments ont été reversées au système de santé par des laboratoires pharmaceutiques pour des traitements anticancéreux dont les résultats n`ont pas été jugés concluants au regard des accords. Plus récemment, un mécanisme de paiement à la performance a été testé, le système de santé italien payant le traitement à l'industriel ex post pour les seuls patients répondeurs. Sources : « Accélération de l'accès à l'innovation pharmaceutique : état des lieux et perspectives », Sophie Joubert, Université d'Angers, 2015 ; « Innovation médicamenteuse en cancérologie, étude internationale sur la définition et l'accès à l'innovation », INCa, janvier 2018. |
2. La France à la traîne de l'Europe ?
Les choix d'organisation différents d'un État membre à l'autre conduisent à observer des variations, parfois importantes, dans les délais de commercialisation de certains médicaments innovants après l'obtention de l'AMM. Vos rapporteurs ont constaté que notre pays souffre à certains égards de cette comparaison.
L'analyse des données disponibles sur les délais de commercialisation des médicaments, entre celles fournies par les acteurs institutionnels et celles émanant des laboratoires est toutefois complexe car elles ne sont pas établies sur la base des mêmes périmètres. Cela plaiderait pour l'élaboration et le suivi dans le temps d'un indicateur commun à l'ensemble des acteurs, établi selon une méthodologie transparente et distinguant éventuellement selon les catégories de médicaments (par exemple en fonction de leur ASMR ou le bénéfice préalable d'une ATU).
On constate dans tous les cas que le délai de 180 jours fixé au niveau européen par la directive « transparence » 43 ( * ) pour les procédures d'inscription sur la liste des médicaments pris en charge par les systèmes d'assurance maladie n'est pas respecté .
• Au niveau institutionnel , ce délai, il faut le regretter, ne fait pas l'objet d'un suivi particulier par le ministère en charge de la santé .
La HAS et le CEPS communiquent des données qui ne permettent pas d'avoir une vision complète et consolidée de l'ensemble du processus.
- D'après la HAS, le temps d'examen des dossiers par la commission de transparence s'établit en moyenne à 105 jours en 2016 et 88 jours en 2017 pour l'évaluation des médicaments en première inscription , soit dans un délai désormais conforme au maximum réglementaire pour la part qui incombe à la phase d'évaluation. Il faut saluer les efforts déployés par la commission de transparence pour réduire ce temps d'évaluation .
- Le rapport d'activité 2016 du CEPS retrace quant à lui le temps moyen de traitement des demandes de première inscription au remboursement des médicaments en ville 44 ( * ) , qui s'établit à 166 jours en 2016 45 ( * ) , dont 284 jours pour les seuls médicaments non génériques ; il oscille, pour ces médicaments, entre 275 jours pour les dossiers ayant abouti à un accord ( 41 jours de plus qu'en 2015 ) et 326 jours pour ceux n'ayant pas abouti à un accord (par abandon, retrait ou rejet).
Pour les dossiers d'inscription de médicaments à l'hôpital , la seule procédure d'examen par le CEPS, après leur évaluation et le cas échéant leur inscription sur la liste en sus pour lesquelles vos rapporteurs n'ont pas eu communication de données spécifiques, est en moyenne de 100 jours en 2016 46 ( * ) (contre 97 jours en 2015), dont 114 jours pour ceux de la liste en sus des prestations d'hospitalisation et 55 jours pour les médicaments rétrocédables.
• Les seules données permettant d'établir une comparaison avec la situation dans les autres pays européens émanent de la fédération européenne des laboratoires pharmaceutiques (EFPIA) 47 ( * ) .
Si certains peuvent en critiquer les biais, force est de constater que ces données sont plus lisibles et davantage appropriées par les acteurs.
Ces données ne correspondent pas, pour la France, aux chiffres des acteurs institutionnels, car elles ne portent ni sur le même périmètre de médicaments ni sur la même période de référence (sont concernés des médicaments ayant obtenu une AMM sur une période considérée, et non les dossiers traités dans l'année par la HAS et le CEPS). Elles ne sont pas superposables avec le délai de 180 jours, en partant notamment de l'octroi de l'AMM et non du dépôt du dossier par l'industriel à la HAS et au CEPS, ce qui peut inclure des délais imputables à l'entreprise.
Ces données apportent néanmoins un éclairage intéressant même si elles appellent quelques nuances : d'une part, elles ne prennent pas en compte les produits ayant fait l'objet en France d'une procédure d'ATU , ce qui contribue à « dégrader » le classement relatif de notre pays ; d'autre part, elles n'éclairent pas pleinement sur l'équité d'accès aux médicaments dans les différents pays, notamment en termes de modalités de prise en charge financière.
D'après cet indicateur 48 ( * ) , la France se positionne relativement loin derrière des pays comme l'Allemagne, le Royaume-Uni ou encore l'Italie, avec un délai médian entre l'AMM et la commercialisation de nouveaux médicaments de 405 jours en France pour ceux ayant obtenu une première AMM entre 2013 et 2015.
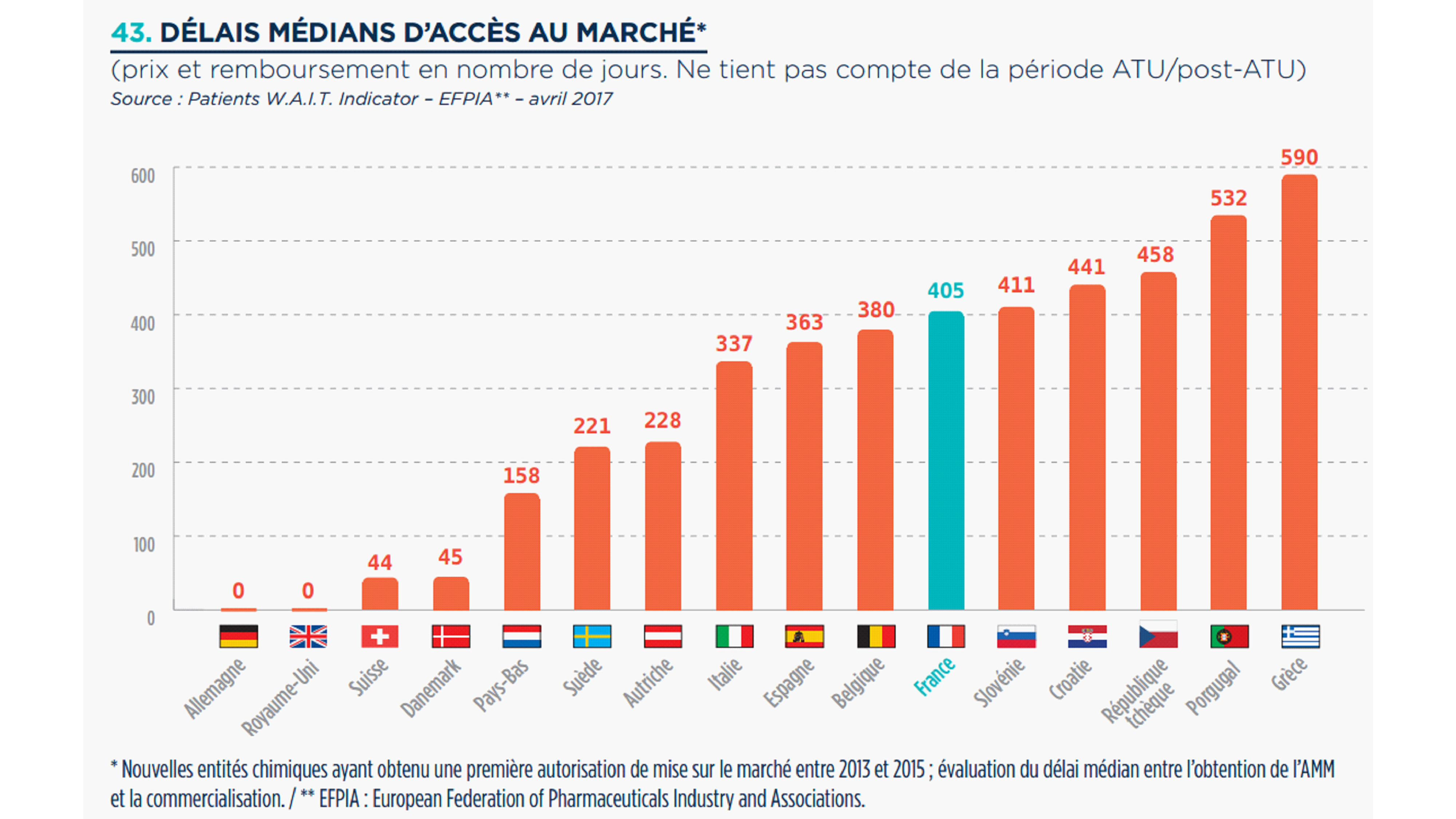
Ce délai médian pour l'accès au marché français est en augmentation puisqu'il était de 363 jours sur la période 2011-2014 et s'établirait à 460 jours sur la période 2014-2016 (d'après des données provisoires).
Le délai moyen passe dans le même temps de 408 jours (sur la base de 139 médicaments ayant obtenu une AMM sur la période 2011-2014) à 530 jours (sur la base de 36 médicaments ayant obtenu une AMM sur la période 2014-2016).
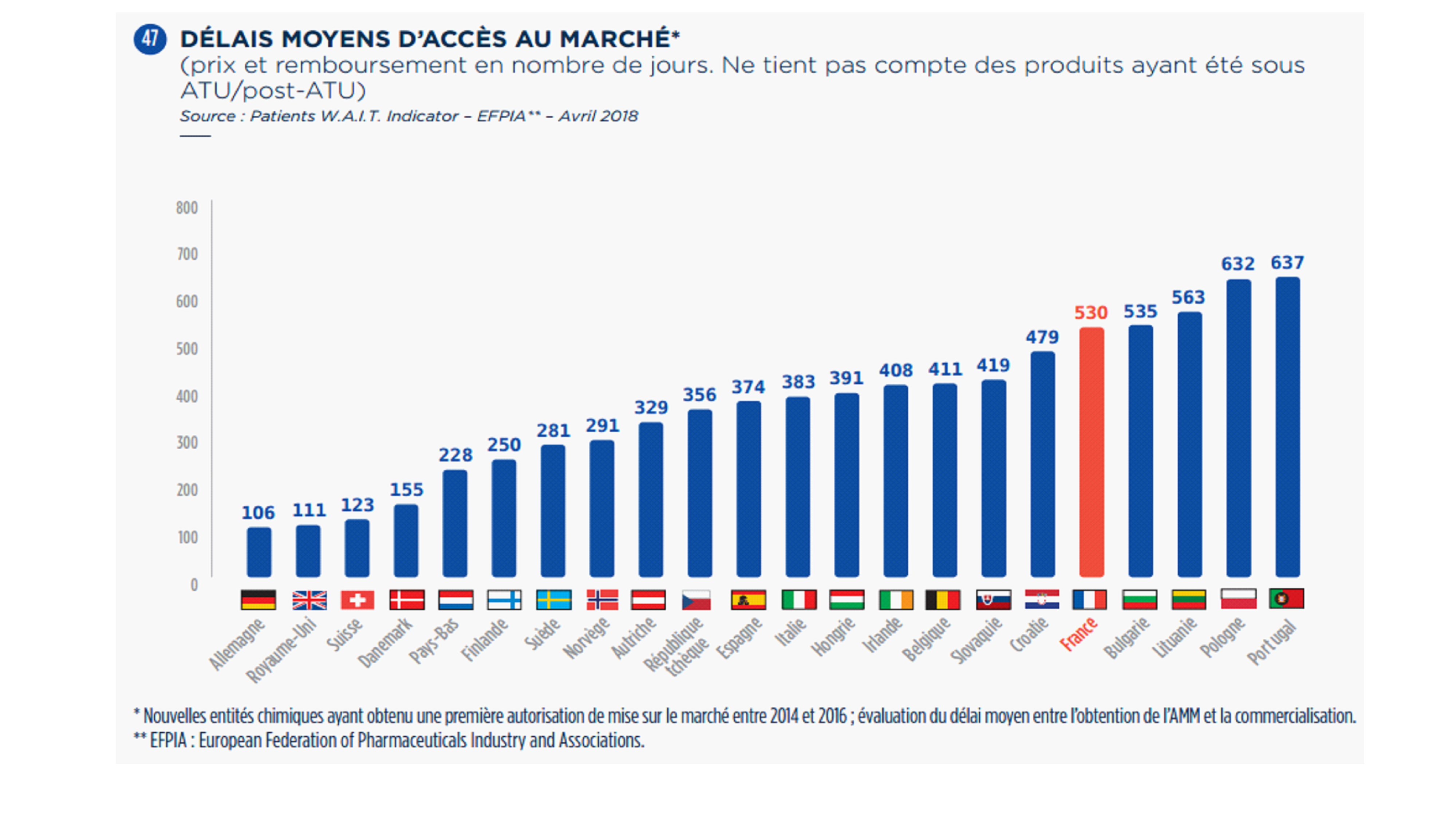
Selon ce même indicateur, le taux de disponibilité des nouveaux médicaments ayant reçu une AMM entre 2011 et 2014 est de 56,6 % en France quand il dépasse les 80 % en Allemagne, en Suisse ou encore en Suède.
• La procédure n'est guère plus rapide pour les médicaments ayant fait au préalable l'objet d'une ATU : une récente étude du Leem ciblée sur 36 produits montre des délais moyens entre l'AMM et la publication du prix au Journal officiel de 389 jours en moyenne, et de 361 jours à partir du dépôt du dossier par l'industriel, soit bien au-delà des 180 jours réglementaires.
Délais moyens et médians de la
procédure d'évaluation
et de fixation du prix de
médicaments ayant fait l'objet d'une ATU
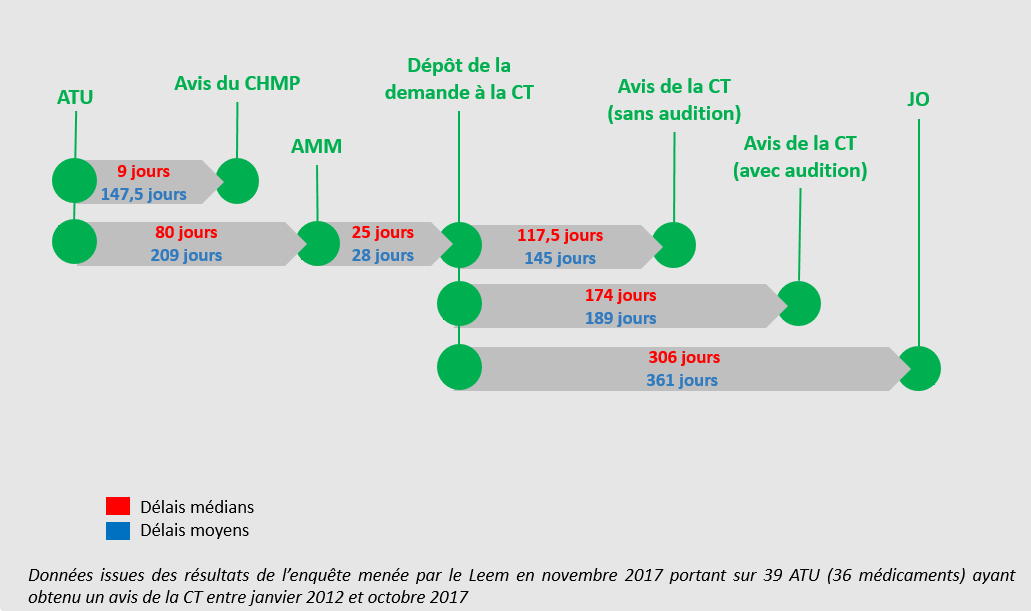
Source : Leem
3. Quels sont les limites et les freins identifiés ?
a) L'évaluation par la HAS : une rigueur reconnue, des modalités réinterrogées dans un contexte d'accélération de l'innovation
Nombre d'interlocuteurs ont souligné les efforts déployés par la HAS pour accélérer ses procédures, accompagner en amont les industriels et permettre un accès plus précoce des patients aux médicaments innovants.
Il faut ainsi souligner que d'après une étude internationale de l'INCa précitée sur l'accès aux innovations médicamenteuses en cancérologie, les agences d'évaluation allemandes et françaises se positionnent en Europe plutôt rapidement par rapport aux autres pays étudiés et sont plus enclines à se positionner positivement.
Toutefois, si la rigueur scientifique de cette procédure et son caractère stratégique sont indéniables, force est de constater que l'accélération des innovations et les enjeux de leur accès précoce conduisent à interroger les modalités actuelles de cette évaluation.
Si cette phase n'est pas perçue comme le frein principal à l'accès précoce des patients français aux molécules innovantes, elle ne va pas sans soulever des interrogations, parfois sources d'incompréhensions : est-elle encore adaptée dans le contexte actuel de l'innovation ?
• Du point de vue de la HAS, le manque de robustesse des données dont la commission de la transparence dispose pour apprécier l'efficacité et la valeur-ajoutée des médicaments présentés comme innovants rend l'évaluation plus complexe .
Cela tient au fait que les AMM sont délivrées de manière de plus en plus précoce par l'EMA, voire à titre conditionnel, sur la base de données jugées immatures (préliminaires, c'est-à-dire en phase 2 des essais cliniques).
Comme l'a relevé sa présidente, ce niveau de preuve conduit la HAS à faire un « double pari » , à la fois sur la réalité de l'efficacité du produit et sa tolérance, incertaines à long terme, mais aussi sur l'impact sur les finances publiques compte tenu des prix revendiqués par les industriels.
• Pour certains industriels, cela conduirait la commission de la transparence à la prudence voire à un excès de prudence dans l'évaluation de l'ASMR de médicaments qu'ils considèrent comme innovants.
Les données disponibles sur l'évolution des ASMR attribuées depuis 2008 ne permettent pas, toutefois, de confirmer une tendance à l'affaiblissement des ASMR I à III correspondant à des innovations importantes ou de rupture qui demeurent extrêmement rares. Le graphe suivant traduit une certaine stabilité, même si le nombre d'ASMR IV est depuis 2015 d'un niveau supérieur aux années précédentes.
Évolution de la répartition des ASMR
attribuées par la HAS
(demandes de première inscription ou
d'inscription
dans une nouvelle indication)
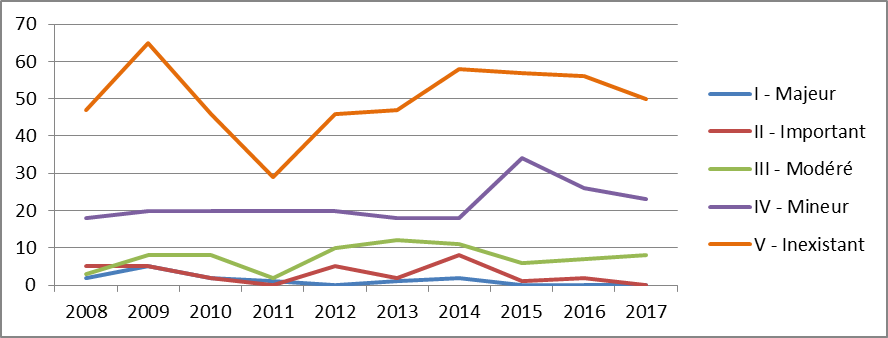
Source : HAS. Ces données ne portent pas sur les demandes d'avis ayant fait l'objet d'une procédure simplifiée.
• D'autres interrogations portent sur l'adaptation de cette procédure aux besoins et à la réalité des thérapies innovantes .
Vos rapporteurs ont ainsi pu constater les incompréhensions que cette procédure peut susciter en particulier chez certaines entreprises de biotechnologies parfois spécialisées sur un produit ou une pathologie :
- l'évaluation ne tiendrait pas suffisamment compte des spécificités des médicaments orphelins liés aux maladies rares : les données portent sur des populations forcément très restreintes et les maladies sont méconnues des experts de la commission de transparence qui peuvent remettre en cause certains critères ou analyses pourtant validés au stade de l'AMM. La prévention des conflits d'intérêt, quelle que soit la légitimité de ces règles et garanties, conduit à ce que cette évaluation ne soit pas réalisée par des spécialistes des pathologies qui seraient jugés plus capables de mesurer l'enjeu de certains nouveaux traitements, a fortiori dans le cas de maladies rares pour lesquelles il n'existe parfois qu'un seul spécialiste en France. En outre, au stade de l'évaluation médico-économique, la méthodologie « standard » de la CEESP ne serait pas jugée adaptée à des études portant sur des populations forcément restreintes ;
- certains comparateurs pris en compte pour l'évaluation de l'ASMR sont jugés contestables , créant là aussi des situations d'incompréhension, par exemple lorsqu'un médicament qui a fait l'objet d'une ATU - et donc, par définition, répond à un besoin médical non couvert - obtient par la suite une ASMR nulle en état comparé à un traitement jugé non pertinent.
Des praticiens hospitaliers ont confirmé le caractère paradoxal, à leur sens, des résultats de l'évaluation de certains traitements qu'ils perçoivent comme efficaces , pouvant parfois conduire à remettre en question leur prise en charge.
• Pour autant, vos rapporteurs saluent, dans ce contexte, les efforts déployés par la commission de la transparence pour réduire les délais d'examen des dossiers, mais aussi pour développer le dialogue en amont avec les industriels : plusieurs outils mis en place par la HAS visent à leur permettre de mieux appréhender ses exigences et donc à accélérer l'évaluation des médicaments présumés innovants.
Si, d'une façon générale, les industriels se saisissent encore peu de ces outils qui manquent selon le Leem d'agilité et de souplesse, certains ont reconnu leur intérêt et la nécessité de les renforcer.
|
Les outils mis en place par la HAS pour
accélérer l'évaluation
• Les « rencontres précoces » peuvent se tenir au niveau national ou européen en amont des demandes d'inscription au remboursement, c'est-à-dire avant les phases III des essais cliniques. L'objectif est d'identifier, dans une phase préliminaire de développement du produit, les points utiles pour l'évaluation future du médicament par la HAS ; il ne s'agit pas toutefois d'une pré-évaluation. Cette mission a été renforcée par la loi de modernisation du système de santé de 2016 pour les produits innovants. 19 rencontres précoces ont été réalisées en 2017. • Un processus d'évaluation anticipée , permettant à l'industriel, pour un médicament présumé innovant, de déposer son dossier auprès de la HAS dès le dépôt de la demande d'AMM , ce qui permet de démarrer de manière anticipée l'instruction du dossier. Il est également possible de déposer un pré-dossier dès l'avis du comité se prononçant sur l'AMM au niveau européen 49 ( * ) . Ce dernier processus n'a été utilisé, d'après les données de la HAS, que pour 31 des 261 nouveaux médicaments évalués entre 2014 et 2017, et pour seulement 22 % des produits en ATU. |
b) Des tensions qui se focalisent sur la phase de négociation du prix
Vos rapporteurs ont constaté, en lien direct avec la question des délais d'accès au coeur de leur réflexion, que c'est sur l'étape de fixation des prix, phase cruciale pour les industriels, que se concentrent les difficultés.
• Si la lettre d'orientation ministérielle de 2016 affiche clairement comme objectif l'accès des patients aux innovations thérapeutiques « dans les meilleurs délais chaque fois que leur état de santé le justifie » , en invitant le président du CEPS à rechercher « le juste prix de ces innovations » à la fois « compatible avec la soutenabilité de la diffusion de ces traitements et un retour sur investissement pour les industriels » , la plupart des acteurs ont relevé un allongement de ces délais en raison d'un durcissement des positions de part et d'autre :
- d'un côté, les exigences financières des industriels sont jugées trop élevées, parfois même extravagantes , notamment au regard de résultats de l'évaluation. Ainsi, pour la Cnam, les promesses des industriels ne sont pas toujours tenues : une analyse du prix des médicaments contre le cancer a ainsi mis en avant « une augmentation régulière et significative des prix par année de vie gagnée » , de 11 % chaque année entre 1995 et 2016 50 ( * ) . Plusieurs campagnes publiques, à l'initiative de Médecins du monde ou de cancérologues ont alerté récemment sur les prix des médicaments et la pression financière exercée par les industriels sur les systèmes de santé.
Certains dénoncent par ailleurs les stratégies des entreprises tendant à se recentrer sur des aires thérapeutiques ciblées à plus fort potentiel économique et à commercialiser des produits d'abord dans une indication visant une population restreinte, pour obtenir un prix plus élevé, avant d'élargir à des indictions visant des populations plus larges ;
- de l'autre, les industriels estiment que l'innovation n'est pas toujours reconnue, valorisée et rémunérée à sa juste valeur . Ce constat émane notamment des entreprises de biotechnologies spécialisées parfois sur un petit nombre de produits. Certains ont estimé que la pression sur les prix était plus forte que dans d'autres pays européens, comme l'Allemagne, avec des prix nets (après remises) bien inférieurs à ceux négociés outre-Rhin, ou encore que le recours à des accords prix-volume conduisait à plafonner rapidement les gains. Les prix nets négociés pays par pays n'étant pas transparents au niveau européen, il n'est toutefois pas possible pour vos rapporteurs d'apprécier la réalité et l'ampleur de ce différentiel 51 ( * ) .
• Pour le Leem, la lettre d'orientation ministérielle de 2016 contribue, plus généralement, à tendre les négociations devant le CEPS et devrait être revue. Dans l'étude précitée de septembre 2017 sur la fixation du prix des médicaments, la Cour des comptes relève en effet que ces lettres d'orientation « sont inspirées de manière croissante par des objectifs de maîtrise du coût des médicaments » et que les orientations de 2016 « marquent un tournant en assignant au président du CEPS des objectifs plus volontaristes et des principes plus rigoureux dans la fixation du prix des médicaments. »
La lettre d'orientation ministérielle de 2016 fixe notamment les principes suivants :
- pour les médicaments sans ASMR (soit ASMR V), le prix doit être « inférieur au prix du comparateur le moins cher » afin de permettre de réaliser une économie dans le coût de traitement ; le CEPS reconnaît que certains produits « font état d'améliorations qualifiées d'incrémentales par l'industriel, qui touchent la forme galénique ou les modalités d'administration dont les impacts hypothétiques en termes d'observance, de qualité de vie ou de coûts d'administration n'ont pas donné lieu à une valorisation au travers de l'ASMR » 52 ( * ) ;
- pour les médicaments avec une ASMR mineure (soit ASMR IV), qui couvrent un champ très large, l'indication concernée « ne doit pas entraîner d'augmentation dans les dépenses de coût de traitement défini par rapport au coût net du comparateur le moins cher pour cette indication », ce qui constitue un durcissement de l'appréciation 53 ( * ) .
Pour le CEPS, cela s'inscrit dans une doctrine récurrente selon laquelle « le bénéfice de l'innovation pour l'entreprise consistera dans l'accroissement de ses parts de marché, sans qu'il y ait lieu d'y ajouter un avantage de prix ».
• Si vos rapporteurs souscrivent aux objectifs de soutenabilité et de nécessaire maîtrise de la dépenses qui sous-tendent ces principes, force est de constater que, dans ce contexte, la phase de négociation suscite des tensions voire des incompréhensions. Comme le reconnaît le CEPS, « le différentiel entre les attentes du laboratoire et les prix résultant de cette approche n'ont pas toujours permis de parvenir à un accord » 54 ( * ) . Vos rapporteurs n'ont pas eu communication toutefois de données globales permettant d'étayer ce constat.
Parmi les critiques exposées par les laboratoires, la question des « comparateurs économiquement pertinents » 55 ( * ) pris en compte, parfois anciens, est à noter : comme pour les comparateurs cliniques intervenant lors de la phase d'évaluation, certains sont contestés dans leur pertinence, a fortiori quand ils s'appliquent à des médicaments ayant fait l'objet préalablement d'une ATU et qui répondent, par définition, à un besoin non couvert.
Au-delà, un laboratoire a récemment manifesté publiquement les tensions dans les négociations de prix devant le CEPS : le laboratoire Vertex, spécialisé dans le traitement de la mucoviscidose, a ainsi fait état, en début d'année 2018, du blocage des discussions pour son produit Orkambi® ayant reçu une ASMR IV alors que le laboratoire s'attendait à une évaluation plus favorable. Cette entreprise a évoqué, dans cette situation d'incertitude, son intention de ne pas débuter des essais cliniques en France pour un nouveau produit. Tout en entendant les arguments des uns et des autres et les rigidités des procédures, vos rapporteurs ne peuvent toutefois adhérer à une position qui conduirait à prendre en otage des patients.
B. INVENTER DES RÉPONSES NOUVELLES POUR MIEUX ANTICIPER, ACCUEILLIR ET ACCOMPAGNER LES INNOVATIONS
1. Développer une vision plus prospective et renouer la confiance entre les acteurs
a) Créer un cadre pérenne d'échanges pour gagner en visibilité
Un certain nombre d'acteurs ont mis en avant un besoin d'améliorer la visibilité sur les innovations à venir, qui s'annoncent pleines d'espoir pour les patients mais dont les coûts potentiellement élevés suscitent des inquiétudes quant à leur impact sur l'assurance maladie.
Confrontés à ce même contexte, plusieurs pays ont mis en place des mécanismes de détection précoce des médicaments innovants , afin que l'ensemble des acteurs du système de santé disposent d'une vision anticipée du marché en amont des AMM, en général un à trois ans avant l'arrivée sur le marché des médicaments : comme le souligne une récente étude de l'INCa, ces dispositifs « partagent l'ambition (...) d'une part d'apprécier et anticiper la charge d'évaluation attendue pour les agences de régulation, et, d'autre part, d'évaluer l'impact potentiel, voire économique, de ces nouveaux médicaments sur le système de santé des pays » 56 ( * ) . Ils existent, sous des formes différentes, en Italie (projet Horizon scanning ), au Royaume-Uni, au Canada ou aux États-Unis.
Les enjeux du 8 ème conseil stratégique des industries de santé (CSIS) qui devrait aboutir à des conclusions et des préconisations début juillet 2018 rejoignent cet objectif. Ses coordonnateurs ont mis l'accent, lors de leur audition, sur la nécessité de mettre en place un « pilotage de l'après » qui serait particulièrement opportun ne serait-ce que pour assurer le suivi dans le temps des propositions qui en émaneront.
Plus généralement, un cadre pérenne de dialogue sur les innovations, associant les entreprises pharmaceutiques et le régulateur (administrations et organismes concernés, assurance maladie...), permettrait d'assurer une meilleure visibilité pour anticiper l'innovation plutôt que la subir, mais aussi développer une vision partagée de ce qu'est l'innovation 57 ( * ) .
Pourrait être développé et suivi dans ce cadre un indicateur sur les délais d'accès au marché des médicaments innovants, alors que des données partagées et consolidées font aujourd'hui défaut.
|
Proposition n° 5 : Structurer un cadre pérenne d'échanges pour anticiper sur les innovations à venir susceptibles d'impacter le système de santé. |
b) Cibler les efforts sur les priorités
Comme l'a souligné la présidente de la HAS, tout ce qui est nouveau n'est pas innovant : pour être innovant, un médicament doit répondre à un besoin thérapeutique en apportant un progrès par rapport à l'existant.
|
Peut-on définir l'innovation médicamenteuse ? Une étude internationale de l'INCa sur l'innovation médicamenteuse en cancérologie, publiée en janvier 2018, a tenté de définir ce qu'est l'innovation et a comparé les procédés permettant de faciliter leur mise à disposition. Un premier constat est qu' une majorité de systèmes ou organismes ne reconnaissent pas de définition officielle à l'innovation médicamenteuse dans les processus d'évaluation : la réticence des autorités sondées à retenir une définition officielle, stable et partagée, vient du caractère restrictif et contraignant qu'aurait une telle approche, l'innovation étant perçue comme « une approche évolutive nécessitant adaptabilité et modularité en fonction des différents médicaments et des différentes pathologies. » Plusieurs systèmes acceptent toutefois d'identifier l'innovation via différents critères objectifs « classiques ». Parmi les exemples de dispositifs mis en place : - le label « Breakthrough therapy » par la FDA aux États-Unis ( Safety and Innovation Act de 2012) vise à accélérer le développement et l'évaluation des médicaments bénéficiant de l'appellation , c'est-à-dire répondant aux critères suivants : le médicament est appelé à traiter, seul ou en combinaison, une maladie grave ou mortelle ; des indicateurs cliniques préliminaires montrent des bénéfices significatifs par rapport à l'existant ; - le système italien identifie le degré d'innovation du médicament , en parallèle au dépôt de dossier pour la fixation du prix et l'admission au remboursement, sur la base de trois critères : gravité des pathologies ciblées ; réponse apportée à un besoin médical non ou mal couvert ; amélioration du service médical rendu. Un critère médico-économique pourrait s'y ajouter. L'étude met en avant des critères objectifs identifiés comme participant à définir l'innovation , parmi lesquels deux se détachent et emportent un niveau élevé de consensus entre les experts de différents pays : - la réponse à un besoin médical non ou mal couvert ; - l' amélioration de survie globale . Viennent ensuite : l'appartenance à une nouvelle classe thérapeutique, l'amélioration de la qualité de vie du patient, la confirmation des résultats en vie réelle, l'augmentation de la durée de rémission... |
Améliorer la prospective sur les innovations thérapeutiques pourrait contribuer à mieux adapter les réponses en fonction des priorités identifiées.
Cela pourrait ainsi conduire à cibler les moyens pour accélérer les procédures d'accès au marché de produits innovants fléchés comme prioritaires en raison du besoin thérapeutique auquel ils permettent de répondre , selon des procédures de « fast track » mises en place dans certains pays, afin de permettre leur commercialisation dans des délais plus rapides après l'AMM.
Cela pourrait concerner en priorité les produits ayant fait préalablement l'objet d'une ATU, sur la base d'un recueil plus efficace des données cliniques tirées de l'utilisation de ces produits au cours de cette phase que vos rapporteurs appellent de leurs voeux ( cf. proposition n° 1) ou ceux susceptibles de bénéficier d'une prise en charge temporaire ( cf. proposition n° 9).
En contrepartie, certaines missions de la HAS pourraient être allégées, en étudiant, suivant la proposition portée par sa présidente, l'opportunité de procéder aux réévaluations tous les dix ans par classe de produit et non tous les cinq ans par produit (hors auto-saisines ou saisines du ministère). Cela ne devrait pas pour autant faire obstacle à la réévaluation plus fréquente des produits les plus innovants.
|
Proposition n° 6 : Envisager une procédure accélérée d'accès au marché (de type « fast-track ») pour des produits innovants fléchés comme prioritaires. |
c) Revoir les critères de l'évaluation pour renforcer leur lisibilité
La nécessité de réformer les critères de l'évaluation du médicament est depuis quelques années bien identifiée.
Comme l'a relevé l'Institut Montaigne dans un récent rapport sur l'innovation en santé, « le besoin de réforme semble acté, mais il doit s'opérer autour des principes de lisibilité, d'efficacité et d'adaptabilité des processus et critères d'évaluation employés par la HAS » 58 ( * ) .
Vos rapporteurs partagent cette observation, d'autant qu'ils ont pu constater un dialogue parfois compliqué entre la HAS et les industriels, sans possibilité pour eux d'engager de recours contre de simples avis 59 ( * ) .
De nombreux représentants des entreprises pharmaceutiques ont en effet considéré que les critères de cette évaluation étaient devenus illisibles et imprévisibles . Cela plaide pour une plus grande formalisation de la doctrine de la commission de la transparence de la HAS, dont les grands axes figurent dans son rapport annuel d'activité.
Au-delà, vos rapporteurs souscrivent au principe d'une réforme que la commission des affaires sociales a déjà appelée de ses voeux : le rapport précité sur la politique du médicament publié en juin 2016 par Gilbert Barbier et Yves Daudigny préconisait ainsi un indicateur unique d'évaluation comparative du médicament, sur la base de l'indice de « valeur thérapeutique relative » envisagé par Dominique Polton dans son rapport de novembre 2015 60 ( * ) ; deux rapporteurs généraux successifs avaient auparavant proposé une réforme en ce sens lors de la discussion au Sénat des projets de loi de financement de la sécurité sociale pour 2014 et 2015.
Par ailleurs, les outils permettant de renforcer le dialogue en amont avec les industriels, comme les « rencontres précoces » décrites plus haut, sont à consolider, ce qui implique que la HAS ait les moyens de cette ambition pour faire preuve de réactivité dans leur mise en oeuvre.
|
Proposition n° 7 : Refonder les critères de l'évaluation du médicament pour renforcer leur lisibilité et leur compréhension. |
d) Développer l'évaluation médico-économique, en tenant compte des spécificités liées aux maladies rares
Lors de leur audition, les représentants des usagers de France Assos Santé ont relayé le sentiment qu'une pathologie commence à coûter cher, aux yeux du régulateur, le jour où arrive un nouveau traitement. Pourtant, l'absence de traitement adapté a également un impact financier pour notre système de soins, parfois non négligeable, de même qu'un impact sociétal. Des professionnels de santé ont également souhaité que soit mieux pris en compte le bénéfice économique ou social à attendre d'un meilleur ciblage des traitements, en particulier dans la prise en charge du cancer.
Si la lettre d'orientation ministérielle d'août 2016 invite le CEPS à prendre en compte, dans la fixation du prix, les « éventuelles économies générées pour l'assurance maladie du fait des bénéfices qu'il apporte ou de son impact organisationnel » , concrètement, les avis de la CEESP ont eu un impact limité de par leur caractère « purement méthodologique » d'après le CEPS. Cela a seulement impacté l'octroi ou non de la garantie de prix européen 61 ( * ) .
Les conclusions du précédent CSIS, en avril 2016, avaient pointé cette limite : « d'une manière générale, les objectifs budgétaires annuels (...) sont difficiles à concilier avec la prise en compte des économies de long terme promises par certains produits de santé ». Force est de constater que les avancées attendues pour objectiver les économies attendues et les prendre en compte dans le prix des produits ont été trop modestes.
Vos rapporteurs estiment que cette évaluation médico-économique devrait prendre une place plus importante sous réserve toutefois que sa finalité, ses critères et modalités soient transparents et partagés entre les acteurs. Elle devrait également rester distincte de l'évaluation de la valeur thérapeutique à laquelle procède la commission de transparence de la HAS.
En particulier, la situation des maladies rares devrait être mieux prise en compte dans la méthodologie de la CEESP . Vos rapporteurs se sont en effet fait l'écho de difficultés spécifiques concernant des médicaments dits « orphelins » impactant un nombre extrêmement réduit de patients, pour lesquels les études ne correspondent pas aux « standards » attendus par cette commission. Or, la formulation de « réserves méthodologiques majeures » impacte ensuite les négociations de prix y compris pour les médicaments les plus innovants. A cette fin, une « cellule » dédiée aux maladies rares pourrait être envisagée au sein de la CEESP.
|
Proposition n° 8 : Développer l'évaluation médico-économique des médicaments innovants, en prenant en compte de manière globale les économies qu'ils sont susceptibles de générer pour le système de santé. Intégrer dans ce cadre les spécificités liées aux médicaments orphelins. |
2. Adapter la grille de lecture au contexte de l'innovation et aux besoins des patients
a) Envisager un processus plus dynamique d'évaluation et de prise en charge des médicaments innovants
L'octroi d'AMM à titre conditionnel ou sur la base de données cliniques préliminaires interroge les procédés « classiques » d'évaluation et les manières de concilier accès précoce et sécurité des patients. Ces évolutions imposent également de nouvelles approches pour gérer l'incertitude de l'impact financier de certains traitements innovants.
Lors de son audition, et suivant le principe de pari éclairé envisagé par Dominique Polton dans son rapport précité sur l'évaluation du médicament, la présidente de la HAS a suggéré de rendre possible, pour des médicaments « prometteurs », la possibilité d'un remboursement temporaire, conditionné à l'apport de data supplémentaires par l'industriel après l'obtention de l'AMM. Ce mécanisme pourrait s'appliquer à des cas précis notamment de pathologies graves et en situation de besoin clinique, pour permettre un accès précoce des patients à des stratégies thérapeutiques potentiellement innovantes - comme demain les CarTcell dans la thérapie génique du cancer ou encore pour des médicaments orphelins - tout en palliant les écarts entre les données disponibles et celles nécessaires pour décider de mesures pérennes de prise en charge.
Cette possibilité d'accorder une prise en charge temporaire des médicaments existe en Grande-Bretagne pour des traitements anticancéreux ou en Allemagne : la G-BA (équivalent allemand de la HAS) décide alors de réviser sa décision à l'issue d'une période définie (qui peut aller d'un à cinq ans), sur la base de données supplémentaires (cliniques ou de vie réelle) collectées par l'industriel 62 ( * ) ; le système des nouvelles méthodes d'examen (NUB) permet en outre aux hôpitaux de négocier le remboursement temporaire de l'innovation, pendant un à trois ans, avant qu'elle ne soit incluse dans le système classique 63 ( * ) .
Les représentants du Leem ont avancé une proposition similaire, fondée sur une évaluation transitoire des innovations et leur prise en charge conditionnelle dans le cadre d'un contrat d'engagement avec les entreprises.
Vos rapporteurs souscrivent à cette idée qui suppose toutefois que soient clairement définies les conditions de prix initial, de renégociation et de réévaluation dans les délais fixés , mais aussi que soit rendu possible le recueil de données cliniques en vie réelle fiables ( cf. ci-après) et qu'il existe une réelle incitation des industriels à procéder à ce recueil.
Le risque, comme certains acteurs l'ont relevé, est en effet que la pression des patients soit forte une fois le produit proposé, ce qui fait peser un enjeu important sur la robustesse des données permettant à la HAS de réévaluer le produit au terme de la période transitoire.
Un « test » à petite échelle pourrait toutefois être envisagé pour mesurer la faisabilité d'un tel dispositif et en affiner le cadrage.
|
Proposition n° 9 : Envisager, pour des médicaments prometteurs mais insuffisamment développés, la possibilité d'un remboursement temporaire, conditionné à l'apport de données supplémentaires après l'autorisation de mise sur le marché (AMM). |
b) Expérimenter de nouveaux modes de fixation des prix
Sans constituer le point de départ des réflexions de vos rapporteurs puisque de récents travaux précités de la commission des affaires sociales ont porté sur le prix du médicament, la question du coût des innovations et de sa soutenabilité pour notre système de santé est évidemment étroitement corrélée aux enjeux de l'accès aux médicaments innovants 64 ( * ) .
La pétition lancée par les cancérologues Dominique Maraninchi et Jean-Paul Vernant en mars 2016 sur les prix des nouveaux traitements anticancéreux ou de la campagne de Médecins du Monde sur le prix des médicaments contre l'hépatite C notamment ont sensibilisé à ces enjeux.
Vos rapporteurs ont pu constater que de nombreux acteurs s'accordent sur la nécessité de faire évoluer les modes « traditionnels » de financement des innovations et de partage des risques.
La nécessité de sortir du cadre annuel des lois de financement de la sécurité sociale constitue un premier objectif : le fonds de financement de l'innovation pharmaceutique vise, sur le fond, à y répondre mais conduit de fait à affaiblir la visibilité sur ces dépenses sorties de l'Ondam. Par ailleurs, il ne perçoit pas de recettes nouvelles puisqu'il est alimenté par des remises précédemment dévolues à la Cnam.
Plus spécifiquement et suivant la même logique que celle d'un remboursement temporel présentée plus haut, le principe de prix plus évolutifs , en fonction de l'efficacité du médicament « en vie réelle » , c'est-à-dire dans la pratique courante, est depuis quelques années au centre de nombreuses réflexions. Une révision plus dynamique des prix et leur ajustement plus fin seraient pour certains susceptibles de faciliter et d'accélérer les accords de prix et donc la mise à disposition aux patients.
L'idée de prix révisés selon l'efficacité des produits en vie réelle a été avancée par le directeur général de la Cnam 65 ( * ) afin de disposer d'un levier de régulation après l'admission d'un médicament au remboursement. Elle l'a également été par des professionnels de santé ou des représentants des patients, partant du constat que l'efficacité d'un traitement, lorsqu'il est prescrit à l'ensemble de la population cible, n'est pas toujours aussi favorable que celle constatée lors des essais cliniques.
Si les expériences passées - à travers les contrats de performance passés entre le CEPS et des industriels 66 ( * ) - ne sont guère jugées concluantes, le rapport récemment remis à la ministre en charge de la santé en mai 2017 67 ( * ) a souligné « l'enjeu majeur » de ces données en vie réelle « pour la qualité des soins et la régulation du système de santé » et la nécessité de développer les outils pour en faciliter le recueil et l'exploitation.
|
Les contrats de performance La possibilité de conclure des contrats de performance est prévue par l'article 12 de l'accord-cadre de 2015 entre le Leem et le CEPS : « Au cas où les modalités de fixation des prix de droit commun ne permettraient pas de trouver un accord, le prix de certains médicaments peut être fixé, à la demande du comité ou de l'entreprise, conditionnellement au résultat de la performance constatée en vie réelle. » L'évaluation de la performance en vie réelle peut aboutir à la réévaluation du prix ou au versement de remises. Entre 2008 et 2015, 13 contrats de performance ont été conclus. Dans l'étude précitée de septembre 2017 sur la fixation du prix du médicament, la Cour des comptes estime leurs résultats « peu probants », les contrats n'ayant pas été dénoués dans les conditions prévues initialement, notamment lorsqu'une baisse importante des tarifs devait intervenir. Pour la Cour, le mécanisme de ces contrats « devrait être inversé de façon à conditionner, non les baisses de prix, mais l'obtention d'un prix élevé à la démonstration du succès en vie réelle. » La lettre d'orientation ministérielle d'août 2016 invite à y recourir pour des médicaments répondant à des besoins thérapeutiques non couverts, lorsque les garanties de bonne exécution paraissent réunies et « sans faire porter le risque financier sur l'assurance maladie » . |
À cet égard, l'expérimentation menée depuis quatre ans par le laboratoire Roche sur des patientes atteintes de cancer du sein (modèle de prix différenciés, en fonction des résultats constatés 68 ( * ) ) mérite l'attention en ce qu'elle vise à adapter les modes de tarification au développement de la médecine personnalisée en oncologie et à l'accélération des évolutions thérapeutiques (extensions d'indications, associations thérapeutiques...).
Sous réserve de l'étude de la faisabilité technique de tels dispositifs, dont vos rapporteurs ne sous-estiment pas la complexité, des mécanismes de fixation des prix plus souples et plus fins (prix différenciés par médicaments selon l'indication ou l'association avec un autre produit, voire par sous-groupes de patients selon la ligne ou la durée du traitement) mériteraient d'être expérimentés, en se limitant dans un premier temps à des champs bien circonscrits.
Les industriels se sont montrés ouverts à ce type d'évolutions, de même qu'à d'autres modes de régulation pour accompagner les innovations dans la durée tels que l'idée d'enveloppe globale et pluriannuelle par pathologie avancée par l'Institut Montaigne dans son récent rapport précité sur l'innovation en santé : cela consiste à intégrer les thérapies innovantes dans l'enveloppe historiquement allouée au traitement d'une pathologie. Pour le Leem, la capacité des industriels dans un tel cadre à « absorber » les chocs d'innovation nécessiterait un alignement du taux de progression des dépenses de médicaments sur le taux de progression de l'Ondam.
|
Proposition n° 10 : Expérimenter, sur des périmètres d'abord restreints, de nouvelles modalités de fixation des prix des médicaments innovants (prix différenciés par indication ou selon l'association avec d'autres produits, prix plus évolutifs en fonction de l'efficacité en vie réelle, enveloppe globale et pluriannuelle par pathologie, etc.) . |
c) Mieux prendre en compte certains besoins spécifiques
Dans certaines situations, notamment en pédiatrie ou dans le traitement des maladies rares, l'absence d'AMM donne lieu au développement de pratiques de prescription hors-AMM .
Comme cela a été relevé à vos rapporteurs lors des auditions, les industriels peuvent ne pas être enclins à conduire des essais cliniques et revendiquer une AMM, compte tenu de la lourdeur et du coût de la procédure, dans certaines indications considérées comme trop peu rentables au regard de la population cible, ou lorsque les médicaments concernés sont anciens et que leur niveau de prix est faible.
L'exemple du salbutamol a été cité par les représentants de France Assos Santé : retiré du marché pour le traitement de l'asthme, cette molécule ancienne fonctionne depuis quelques années pour des malades atteints d'une maladie rare (le syndrôme myosthémique) pour lesquels il apporte un réel progrès thérapeutique et constitue donc une innovation. Ce médicament est prescrit à ces patients dans le cadre d'ATU nominatives depuis trois ans, le coût d'une AMM n'étant pas rentable pour le laboratoire qui le commercialise.
En outre, comme l'ont relevé les représentants de l'INCa, l'AMM peut être pointue et plus restreinte dans ses indications que l'ATU , en raison notamment de la volonté des industriels de « sécuriser » le dossier de demande d'AMM en ciblant celle-ci sur les indications dans lesquelles les résultats des essais sont les plus solides. La prescription hors AMM peut alors devenir indispensable pour répondre aux besoins de certains patients.
Si le dispositif des RTU permet d'encadrer et sécuriser les pratiques de prescription hors-AMM 69 ( * ) , celui-ci est jugé, comme vos rapporteurs l'ont souligné 70 ( * ) , insuffisamment souple et réactif pour répondre à l'ensemble des besoins.
Vos rapporteurs soulignent l'intérêt, dans ce cadre, de la proposition avancée par l'INCa visant à réguler les prescriptions hors AMM sur la base de référentiels de bon usage nationaux (RBUN), établis sur une base scientifique et clinique et régulièrement actualisés 71 ( * ) .
Pour l'INCa, ces référentiels énonceraient à la fois les prescriptions acceptables et celles qui ne sont pas ou plus justifiées, y compris relevant de l'AMM. Cela permettrait d'harmoniser les pratiques au niveau national et d'offrir aux patients, de façon réactive, les thérapeutiques les plus adaptées selon les données scientifiques disponibles. L'INCa évalue le renfort de ces équipes nécessaires à la mise à jour de ces référentiels, pour ce qui concerne l'oncologie, à deux équivalents temps plein.
Vos rapporteurs notent l'intérêt de cette proposition en terme également de pertinence des prescriptions.
Compte tenu des moyens qui seraient nécessaires pour étendre ce dispositif à l'ensemble des prescriptions, il serait raisonnable d'envisager d'abord une expérimentation sur un champ thérapeutique limité , en priorité en oncologie en se fondant sur l'expertise de l'INCa.
|
Proposition n° 11 : Améliorer le cadre de régulation des prescriptions hors-AMM, sur la base de référentiels nationaux de bon usage, pour sécuriser et rendre plus réactif l'accès aux thérapeutiques adaptées en particulier pour des maladies rares ou certaines situations « de niche ». |
III. GARANTIR L'ÉQUITÉ D'ACCÈS AUX TRAITEMENTS INNOVANTS
L'accès précoce au marché des médicaments n'a de sens que s'il s'inscrit dans une prise en charge solidaire et équitable pour chaque patient, en tout point du territoire.
Face à ce principe fort de notre système de soins se pose, de manière globale, la question de la soutenabilité de la prise en charge des innovations les plus coûteuses.
Plus spécifiquement, vos rapporteurs souhaitent mettre l'accent sur deux outils au rôle stratégique pour l'accès équitable aux médicaments onéreux à l'hôpital d'une part, et pour la diffusion des actes attachés à la montée en puissance de la médecine personnalisée d'autre part.
A. LES MÉDICAMENTS ONÉREUX À L'HÔPITAL : RÉFORMER UN MODE DE PRISE EN CHARGE AUJOURD'HUI FRAGILISÉ
1. La « liste en sus » : un outil indispensable pour assurer l'accès des patients aux progrès thérapeutiques à l'hôpital
a) Un financement dérogatoire
Le financement des médicaments administrés aux patients lors d'un séjour en établissement de santé est assuré selon plusieurs modalités. Dans le cas général, le coût des médicaments est intégré dans le tarif des prestations d'hospitalisation, à savoir dans les GHS (groupes homogènes de séjours).
|
Les modes de financement des médicaments
A l'hôpital, coexistent quatre modes de financement des médicaments : • Les groupes homogènes de séjour (GHS) ( budget Ondam hospitalier) • L'enveloppe MIGAC dont une partie finance les ATU ( budget Ondam hospitalier) • La rétrocession pour les médicaments destinés aux patients en ambulatoire et délivrés par la Pharmacie à Usage Intérieur (PUI) ( budget Ondam ville ) • La liste en sus pour certains médicaments innovants et onéreux, dont l'utilisation chez les patients est trop hétérogène pour qu'ils soient intégrés au sein d'un GHS ( budget Ondam hospitalier ) |
Un dispositif dérogatoire a été prévu, parallèlement à la mise en place de la tarification à l'activité (T2A), pour la prise en charge des médicaments innovants les plus onéreux : instituée par la loi de financement de la sécurité sociale pour 2004, la « liste en sus » permet aux établissements de santé de facturer à l'assurance maladie certaines spécialités pharmaceutiques en sus des tarifs des séjours hospitaliers (ou GHS) et d'obtenir leur remboursement à 100 %.
Ce mode de financement est perçu comme essentiel à la diffusion des traitements innovants et onéreux à l'hôpital , qui sans cela ne seraient pas prescrits dès lors que leur coût est trop élevé pour qu'ils soient intégrés dans le forfait de GHS ou pris en charge sur les fonds propres de l'établissement. Les représentants des fédérations hospitalières ont souligné l'intérêt majeur de ce dispositif afin d'éviter toute perte de chance pour les patients et des ruptures d'équité dans la prise en charge des soins.
b) Une dépense dynamique
En 2016, les dépenses de médicaments de la liste en sus se sont établies à 3 milliards d'euros , en croissance de 23 % depuis 2009.
A titre de comparaison, les médicaments financés au titre des tarifs des prestations d'hospitalisation (GHS) représentent la même année une dépense de 1,8 milliard d'euros 72 ( * ) .
Évolution des dépenses de la liste en sus (médicaments) depuis 2009
(en milliards d'euros)
|
Année |
2009 |
2010 |
2011 |
2012 |
2013 |
2014 |
2015 |
2016 |
|
Dépenses |
2,437 |
2,685 |
2,575 |
2,523 |
2,713 |
2,839 |
2,975 |
3,008 |
|
Evol. (%) |
- |
+ 10,2 |
- 4,1 |
- 2,0 |
+ 7,5 |
+ 4,6 |
+ 4,8 |
+ 1,1 |
Source : DGOS - données ATIH
En 2016, près de 55 % de la dépense totale de médicaments de la liste en sus vient des produits utilisés dans le traitement du cancer ; la part des immunosuppresseurs 73 ( * ) en représente le cinquième (21,3 % des dépenses). Quinze spécialités concentrent à elles seules plus de 79 % de la dépense.
2. Un fonctionnement peu satisfaisant
L'évolution dans le temps de la liste en sus révèle des limites et des dysfonctionnements qui sont très largement relevés, non seulement par les industriels, patients ou professionnels de santé, mais également par les administrations concernées. Ces limites tiennent notamment à des critères de gestion contestés ainsi qu'au caractère sédimenté de cette liste, ne lui permettant pas de jouer correctement son rôle de diffusion des innovations dans le cadre budgétaire contraint de l'Ondam.
a) Une liste sédimentée ?
Certains intervenants ont considéré que la liste en sus était, par certains aspects, « un panier dans lequel on entre et dont on ne sort jamais », créant ainsi des « rentes de situation » pour des molécules n'ayant plus le statut de réelles innovations mais dont le prix demeure, par ce biais, élevé.
Ce constat traduit le caractère jugé globalement peu satisfaisant du mode de régulation de cette liste même s'il appelle quelques nuances.
Afin de permettre une gestion plus fine de la liste en sus, la loi de financement de la sécurité sociale (LFSS) pour 2014 74 ( * ) a permis une première avancée en restreignant l' inscription sur cette liste par indication thérapeutique, et non plus par produit. Néanmoins, les directions compétentes du ministère en charge de la santé reconnaissent à cette évolution certaines limites, dues à la complexité des radiations partielles dans la mesure où il n'est pas possible de financer un même médicament selon différents modes de financement, et à l'absence, jusqu'à une date récente, d'outils permettant un codage par indication et donc un suivi précis des prescriptions.
D'après les données transmises à vos rapporteurs par la direction générale de l'offre de soins, la liste en sus comporte 6 médicaments inscrits depuis plus de 10 ans , sur un total d'environ 400 couples indications-médicaments . 31 radiations sont intervenues depuis la publication du décret du 24 mars 2016 (à fin janvier 2018) ; 81 inscriptions ont été accordées en 2017, pour 10 refus d'inscriptions.
b) Des critères de gestion contestés
En particulier, les critères de gestion de la liste en sus (inscriptions et radiations), fixés depuis 2016 par décret alors qu'ils relevaient jusqu'alors de recommandations 75 ( * ) , et appliqués de manière quasi-automatique, sont jugés trop rigides ou pas toujours en phase avec les avancées de la science ou les besoins des malades .
|
Les critères pour l'inscription et la radiation
Le décret n° 2016-349 du 24 mars 2016 a fixé la procédure et les conditions d'inscription sur la liste en sus, qui faisaient auparavant l'objet d'une simple recommandation. - Les modalités d'inscription L'inscription d'une spécialité pharmaceutique sur la liste en sus - pour une ou plusieurs indications thérapeutiques - est sollicitée par l'entreprise exploitant cette spécialité auprès des ministres chargés de la santé et de la sécurité sociale, ou initiée par ces ministres. Le dossier comporte des éléments d'impact financier. - Les conditions d'inscription Quatre critères cumulatifs doivent être remplis : 1° La spécialité est susceptible d'être administrée majoritairement à l'hôpital ; 2° Le niveau de service médical rendu (SMR) est majeur ou important ; 3° Le niveau d'amélioration du service médical rendu (ASMR) est majeur (I), important (II) ou modéré (III). Il peut être mineur (IV) si l'indication présente un intérêt de santé publique et en l'absence de comparateur pertinent. Il peut être mineur (IV) ou inexistant (V) si les comparateurs pertinents sont déjà inscrits ; 4° Le coût du produit n'est pas compatible avec les tarifs des séjours concernés : le coût moyen estimé du traitement par hospitalisation est supérieur d'au moins 30 % aux tarifs de la majorité des prestations dans lesquelles la spécialité est susceptible d'être administrée. - Les conditions de radiation La radiation est soit demandée par l'entreprise exploitant la spécialité soit prise à l'initiative des ministres chargés de la santé et de la sécurité sociale, dans l'un des cas suivants : 1° L'un des quatre critères d'inscription n'est plus rempli ; 2° La spécialité est administrée dans au moins 80 % des hospitalisations pour les prestations représentant 80 % des administrations de la spécialité dans l'indication ; 3° La ou les indications de la spécialité entraînent des dépenses injustifiées. |
En particulier, le critère d'inscription fondé sur l'évaluation de l'amélioration du service rendu (ASMR) du médicament apparaît comme non pertinent et dévoyé de son objectif initial . La présidente de la HAS reconnaît que l'ASMR, critère médical qui sert de base à la négociation du prix, n'a pas vocation à déterminer un mode de prise en charge ou un critère d'accès au remboursement.
Plusieurs difficultés, même si elles se focalisent en pratique sur un nombre restreint de situations et de patients, sont relevées ( cf. encadré suivant), concernant principalement des produits avec une amélioration du service rendu mineure (ASMR IV), pour lesquels l'inscription sur la liste en sus n'est pas automatique contrairement à ceux présentant une amélioration du service médical rendu majeure, importante ou modérée (ASMR I, II ou III). Dans le cas d'une ASMR IV ou V, un « comparateur pertinent » doit déjà figurer sur la liste en sus , ce qui peut donner lieu à des appréciations divergentes. Au final, des traitements susceptibles non seulement d'après les laboratoires les commercialisant mais également d'après la communauté médicale ou scientifique de présenter un intérêt thérapeutique pour certains malades n'obtiennent pas leur inscription sur la liste en sus.
Comme l'ont relevé les représentants du Leem, certains produits ne peuvent ainsi pas entrer dans la liste en sus pour des questions juridiques et ne peuvent entrer dans les GHS pour des raisons économiques .
Dans ce « vide », nombre d'interlocuteurs ont vu une source d' iniquité entre les établissements de santé, entre les territoires mais aussi entre les patients , selon l'établissement dans lequel ils sont hospitalisés (puisque certains peuvent accepter, sur des populations cibles très restreintes, de prendre en charge des traitements sur leurs fonds propres), ou selon leur mode de prise en charge, en ville ou à l'hôpital, et même parfois selon la sous-population à laquelle ils appartiennent ou la mutation dont ils sont porteurs quand l'évaluation du produit a été différente.
Cette situation est jugée, en outre, discriminante pour les médicaments hospitaliers au regard des produits de ville dont l'admission au remboursement ne dépend pas du niveau d'ASMR mais du SMR.
|
La liste en sus, un fonctionnement contesté Plusieurs exemples de non inscription ou de radiation de produits de la liste en sus ont été récemment contestés - et parfois relayés par les medias - par des professionnels hospitaliers, sociétés savantes ou associations de patients, pour être susceptibles d'entraîner des pertes de chance pour des malades. Au-delà de l'émotion légitime suscitée par des décisions de ce type qui sont toujours difficiles, et sans se prononcer sur des considérations d'ordre thérapeutique, vos rapporteurs estiment que ces exemples illustrent à tout le moins les dysfonctionnements qui leur ont été soulignés. Dans le traitement du mélanome , plusieurs organismes ou associations de patients ou le professeur Pascal Joly, président de la société française de dermatologie, ont alerté sur la radiation de la liste en sus, par un arrêté du 20 novembre 2017, de Yervoy® ( ipilimumab ) 76 ( * ) , évalué avec un SMR insuffisant et sans ASMR. Or, l'utilisation de ce médicament en association avec une autre immunothérapie (Opdivo®, nivolumab ) a été validée par l'INCa comme traitement de première ligne dans certains cas de mélanome métastatique. Cela induit une discrimination entre patients selon le type de mutation dont ils sont porteurs, puisque dans un cas ils peuvent bénéficier d'un traitement par le circuit de prise en charge en ville et dans l'autre ils n'auront plus accès à un traitement à l'hôpital. Dans le traitement du cancer de la prostate métastatique, des patients ont contesté l'absence d'inscription sur la liste en sus, pour des raisons similaires (ASMR mineure et absence des comparateurs de la liste), du Xofigo® (à base de radium 223), financé sur les tarifs des prestations d'hospitalisation et donc non remboursé à l'euro-l'euro comme ceux inscrits sur la liste en sus. • L'INCa et Unicancer ont relevé d'autres exemples de couples produits/indications dans la même situation , parmi lesquels : Avastin® (carcinome du col de l'uterus), Erwinase® (leucémie aigüe lymphoblastique), Yondélis® (sarcomes des tissus mous), Abraxane® (cancer du pancréas), Cyramza® (cancers gastriques). • Dans le domaine de la greffe de rein , des associations de patients 77 ( * ) et praticiens ont contesté, de même, l'absence d'inscription sur la liste en sus d'un antirejet ( belatacept ) présentant des résultats jugés supérieurs aux autres traitements et pris en charge dans de nombreux pays européens. |
3. Réformer la liste en sus pour consolider son rôle dans l'accès équitable des patients aux médicaments innovants
a) Assouplir les critères d'inscription
Les observations ci-dessus invitent à s'interroger sur un assouplissement ou plus largement une révision des critères d'inscription sur la liste en sus .
En particulier, une déconnexion de ces critères de l'ASMR devrait être engagée en liaison avec la réforme des critères d'évaluation du médicament envisagée par le Gouvernement. Cela permettrait notamment de déverrouiller l'accès à la liste en sus pour des médicaments avec aujourd'hui une ASMR IV , afin de mieux s'adapter à la réalité de l'innovation, qui repose non seulement sur des progrès de rupture mais également sur des progrès incrémentaux.
Certains laboratoires pharmaceutiques prônent un critère strictement économique. La piste de réflexion avancée par l'INCa - à savoir un critère fondé sur l'« efficience ajoutée » - est en outre intéressante.
|
Proposition n° 12 : Revoir les critères d'inscription et de radiation sur la liste en sus afin de déverrouiller l'accès pour les médicaments avec une ASMR IV. |
b) Responsabiliser les prescripteurs
La contrepartie d'une plus large ouverture de la liste en sus devrait être une plus forte responsabilisation des prescripteurs , par un suivi plus fin du recours aux produits inscrits sur cette liste .
Il est en effet impératif que la prescription de ces médicaments onéreux soit réservée à des produits qui apportent, pour un patient donné, une amélioration significative par rapport à ceux pris en charge par les tarifs hospitaliers. Or, certains intervenants ont relevé des biais possibles puisque le mode de prise en charge des produits sur la liste en sus peut constituer, en lui-même, une incitation à y recourir de manière privilégiée par rapport à ceux intégrés dans les GHS.
Ces enjeux rejoignent plus globalement ceux sur la pertinence des soins mis en avant par notre rapporteur général dans un récent rapport de la Mecss 78 ( * ) et qui constitue l'un des cinq chantiers de la stratégie de transformation de notre système de santé engagée par le Premier ministre et la ministre en charge de la santé le 13 février 2018.
A cette fin, l'article 51 de la loi de financement de la sécurité sociale pour 2018 79 ( * ) , dont votre commission des affaires sociales a soutenu le principe lors de l'examen de ce projet de loi de financement, permettrait d' envisager, à titre expérimental, un mécanisme d'intéressement à la juste prescription. Cet article a créé un cadre général d'expérimentations visant d'une part à « permettre l'émergence d'organisations innovantes » pour améliorer le parcours des patients et d'autre part à « améliorer la pertinence de la prise en charge par l'assurance maladie des médicaments (...) et la qualité des prescriptions » 80 ( * ) . Ce dernier objectif inclut spécifiquement une modification des conditions de prise en charge des médicaments onéreux au sein des établissements de santé notamment pour inciter à une meilleure régulation de la liste en sus .
|
Proposition n° 13 : Responsabiliser en parallèle les prescripteurs en créant à titre expérimental un mécanisme d'intéressement à la juste prescription des produits figurant sur la liste en sus. |
c) Anticiper les sorties de la liste en sus pour en permettre une gestion plus dynamique
En tant que mode de financement dérogatoire des produits innovants et coûteux, la liste en sus, par définition, doit reposer sur une gestion dynamique afin de pouvoir intégrer de nouveaux produits au fil de leur arrivée sur le marché.
Or, les sorties de cette liste sont aujourd'hui difficiles dès lors qu'elles ne sont pas suffisamment anticipées.
Une anticipation de cette sortie, en liaison avec les industriels, pourrait donc constituer le corollaire indispensable d'une plus large ouverture de la liste en sus, par exemple en programmant une baisse des prix selon un calendrier déterminé, voire en examinant parallèlement la possibilité de revaloriser les GHS pour y intégrer certains produits.
Ces éléments devraient être intégrés dès les négociations de prix avec le CEPS et corrélés à la réévaluation régulière et systématique de la valeur thérapeutique de ces produits par la HAS.
|
Proposition n° 14 : Anticiper les sorties de liste en sus en programmant une baisse des prix voire une revalorisation plus régulière des GHS. |
B. PRENDRE EN COMPTE LES ENJEUX ÉMERGENTS RELATIFS AU FINANCEMENT DES ACTES DE BIOLOGIE INNOVANTS
1. Les enjeux liés au développement de la médecine personnalisée
L'attention de vos rapporteurs a été appelée sur les enjeux liés à la prise en charge des actes de biologie innovants.
En effet, tant les représentants des fédérations hospitalières et de l'INCa que les praticiens rencontrés à l'hôpital Gustave Roussy ont insisté sur le rôle décisif du diagnostic dans le développement de la médecine personnalisée et de précision en oncologie : les tests dits « compagnons » permettent de mesurer des biomarqueurs sur les tumeurs afin de mieux cibler les traitements des patients atteints de cancer en fonction des anomalies moléculaires identifiées et d'améliorer leur suivi.
Cet enjeu est stratégique en termes de pertinence des soins puisque le ciblage permet des « désescalades thérapeutiques » : ce sont à la fois des économies pour le système de santé et une amélioration de la qualité de vie des malades atteints de cancer.
Dans d'autres cas, c'est la tolérance du traitement voire la vie du patient qui sont engagées : certaines molécules entrant dans la composition de 60% des chimiothérapies - les fluoropyrimidines telles que le 5-Fluorouracile (5-FU) ou ses dérivés 81 ( * ) - présentent, comme l'a reconnu l'ANSM en février 2018, un risque élevé de toxicité pour certains patients 82 ( * ) , susceptibles d'entraîner une hospitalisation. Le dépistage en amont des patients exposés à ces risques devient alors essentiel.
2. Les limites de la prise en charge dans le cadre du référentiel des actes innovants hors nomenclature (RIHN)
Or, les modalités de prise en charge financière de ces tests demeurent insatisfaisantes .
Les tests « compagnons » ne sont pas inscrits à la nomenclature. Leur prise en charge relève d'un outil dérogatoire : le référentiel des actes innovants hors nomenclature (RIHN) et sa liste complémentaire .
Créés en 2015 83 ( * ) , ces outils sont en quelque sorte le pendant de la liste en sus pour la prise en charge des actes innovants de biologie médicale et d'anatomocytopathologie dans l'attente de leur évaluation par la HAS et de leur inscription à la nomenclature pour leur prise en charge par l'Assurance maladie dans les conditions de droit commun.
Une enveloppe fermée de l'Ondam hospitalier d'un montant annuel de 380 millions d'euros est dédiée au financement des actes inscrits au RIHN et sur sa liste complémentaire, au titre des dotations MERRI 84 ( * ) .
Toutefois, cette enveloppe ne permet pas de financer l'ensemble des actes réalisés par les établissements de santé .
Pour l'Institut Gustave Roussy, seulement 75 % environ de la valorisation théorique des actes est effectivement perçue ; les fédérations hospitalières ont indiqué que la prise en charge de certains actes de la liste complémentaire dont l'utilité est pourtant reconnue n'est que de 25 %. Ce risque financier conduit à des pratiques différenciées entre établissements et donc à des iniquités entre patients : les représentants de l'Institut Curie ont indiqué ne pas pouvoir prendre en charge, par exemple, des patients adressés par d'autres établissements.
Pour les professionnels entendus par vos rapporteurs, cette situation est indéniablement un frein au développement de certains tests comme les tests compagnon 85 ( * ) .
Valorisation des actes inscrits au RIHN ou sur sa liste complémentaire réalisés
(en millions d'euros)
|
2016 |
2017 |
|
|
RIHN |
245,2 |
349,2 |
|
Liste complémentaire |
421,8 |
408,4 |
|
Total |
667,0 |
757,6 |
Source : DGOS - données remontées par les établissements de santé
3. Fluidifier la gestion du RIHN en accélérant la procédure d'inscription des actes à la nomenclature
Comme pour la liste en sus, une gestion plus dynamique du RIHN et de sa liste complémentaire est indispensable pour éviter les effets de « goulots d'étranglement » limitant l'inscription et la prise en charge de nouveaux actes et donc l'accès des patients aux tests diagnostiques.
Cela vaut d'autant plus pour la « liste complémentaire » qui comporte des actes ayant fait leur preuve - en étant identifiés en 2015 comme d'utilité clinique démontrée - et dont l'inscription à la nomenclature aurait dû intervenir dans un délai d'un an lors de la création de cet outil.
Des professionnels entendus par vos rapporteurs ont souhaité un déblocage rapide de l'inscription de ces actes à la nomenclature . Cette procédure implique l'assurance maladie et la HAS, dont l'avis entraîne la hiérarchisation et la tarification de l'acte par l'Uncam ; seule la Cnam, les sociétés savantes et professionnels de santé peuvent saisir la HAS.
Plus globalement, une meilleure coordination des processus d'évaluation et de remboursement des molécules et des tests « compagnon » qui leur sont liées serait à encourager pour accélérer ce processus.
Cette évolution devrait s'accompagner, comme pour la liste en sus , d'un suivi de la pertinence des prescriptions , que ce soit pour les actes figurant au RIHN ou, plus encore, ceux inscrits à la nomenclature, pour ne pas aboutir à une inflation des prescriptions et maîtriser l'impact budgétaire, et d'une réévaluation régulière des tests pour supprimer de la nomenclature ou du RIHN ceux qui ne sont plus pertinents.
A cet égard, l'initiative de l'Institut Gustave Roussy de création d'un comité de bon usage des biomarqueurs est tout à fait intéressante en vue d'assurer une forme de régulation pour accompagner le passage des biomarqueurs innovants en tests de routine.
|
Proposition n° 15 : Accélérer et fluidifier les procédures d'inscription des actes de biologie innovants à la nomenclature. |
IV. CONFORTER LE RÔLE DES ESSAIS CLINIQUES DANS L'ACCÈS PRÉCOCE DES PATIENTS AUX TRAITEMENTS INNOVANTS
Les essais cliniques précoces, dont le nombre connaît une augmentation régulière dans le monde , sont devenus l'un des moteurs essentiels des innovations thérapeutiques et font désormais souvent partie intégrante des soins. C'est le cas en particulier dans le domaine de la cancérologie, où d'importants besoins médicaux demeurent insatisfaits et où l'avènement de la médecine de précision et la révolution de l'immunothérapie ont ouvert de nouvelles perspectives pour les patients, les soignants et les chercheurs.
Dans ce contexte, la recherche implique une gestion d'études de plus en plus complexes dans un contexte international devenu extrêmement compétitif . Si l'expertise de nos chercheurs est unanimement reconnue et des initiatives structurantes ont vu le jour, la nécessité d'améliorer l'environnement de la recherche clinique française, notamment précoce, par l'identification de priorités stratégiques, une interaction plus étroite de tous les acteurs et le soutien au développement des infrastructures est un constat partagé de tous. Au-delà des enjeux majeurs de financement de la recherche, que le présent rapport n'a pas vocation à aborder, de nombreux freins organisationnels et administratifs doivent en effet être levés pour garantir l'attractivité de la France sans remettre en cause les garanties de sécurité et de déontologie au respect desquelles il convient en toute circonstance de s'astreindre.
A. LA RECHERCHE CLINIQUE COMME VOIE D'ACCÈS PRÉCOCE AUX TRAITEMENTS INNOVANTS
1. Un encadrement juridique étroit, qui a pour principal objectif d'assurer la sécurité des volontaires
a) Les essais cliniques, une étape prometteuse mais encore très incertaine dans le processus de développement d'un médicament
La commercialisation d'une ou de plusieurs molécules sous la forme d'un médicament à usage humain nécessite la mise au jour préalable de connaissances biologiques et médicales acquises dans le cadre d'une recherche biomédicale réalisée sur l'homme. Celle-ci se concrétise par la mise en place d'un essai clinique permettant de tester un médicament « candidat » ou une nouvelle façon d'utiliser un traitement connu. Il s'agit d'établir ou de vérifier, selon la classification opérée à l'article R. 1121-1-1 du code de la santé publique, les données pharmacocinétiques (absorption, distribution, métabolisme et excrétion), pharmacodynamiques (mécanisme d'action) et thérapeutiques (efficacité et tolérance) d'un médicament.
Les essais cliniques s'inscrivent ainsi dans le prolongement d'une phase initiale de recherche exploratoire permettant d'identifier une molécule susceptible de répondre à un besoin médical non pourvu ou d'améliorer les prises en charge existantes, suivie d'une phase obligatoire de recherche préclinique en laboratoire sur des cellules in vitro et/ou sur des animaux. Cette dernière vise à évaluer l'innocuité et l'absence de toxicité de la substance testée. Elle permet en outre de déterminer les doses qui seront administrées à l'homme au cours des essais cliniques.
Une fois ces deux premières phases achevées, la molécule sélectionnée est brevetée et s'ouvre la phase de recherche clinique et industrielle. Réalisés soit chez le volontaire malade, soit chez le volontaire sain, les essais cliniques se décomposent en trois, voire quatre phases successives . Au cours de la phase I, la molécule est évaluée sur un nombre limité de volontaires sains dans l'objectif de déterminer sa tolérance et sa sécurité d'emploi et d'évaluer son évolution dans l'organisme. La phase II consiste à administrer la substance à des patients malades afin d'identifier les doses auxquelles elle est efficace et tolérée. La réalisation d'essais cliniques à plus grande échelle est conditionnée à la preuve de concept établie au cours de cette deuxième phase. Au cours de la phase III, la molécule est administrée à une population significativement plus nombreuse, avec l'objectif de valider à la fois son efficacité et sa tolérance à plus grande échelle et son rapport bénéfice-risque. Si les résultats s'avèrent concluants, le promoteur a la possibilité d'introduire une demande d'AMM en vue d'une éventuelle commercialisation du produit. Enfin, le suivi du médicament en vie réelle se déroule en phase IV dans le cadre de la pharmacovigilance dont l'objectif est d'identifier tout effet secondaire grave et/ou inattendu du médicament.
Les quatre phases des essais cliniques
|
Nombre
|
Durée indicative |
Objectif |
|
|
Phase I |
20-100 |
De plusieurs mois
|
Sécurité et tolérance |
|
Phase II |
jusqu'à 100 |
De plusieurs mois
|
Tolérance à court terme
|
|
Phase III |
100 à plus de 1 000 |
De 1 à 4 ans |
Évaluation et comparaison bénéfice/risque |
|
Phase IV |
Plus de 1 000 |
De 1 à 4 ans |
Tolérance et recherche
|
Source : Ligue nationale contre le cancer
Au total, pour un médicament finalement commercialisé, en fonction des pathologies concernées et du nombre de volontaires participant, environ cinq années pourront s'être écoulées du début de la recherche pharmaceutique à la fin de la phase préclinique et dix au terme de la recherche clinique. Seul un nombre relativement très faible de molécules sont développées jusqu'au terme de ce processus.
b) Un régime juridique contraignant pour protéger les personnes qui acceptent de se prêter à une recherche
La réalisation d'un essai clinique étant empreinte de nombreuses inconnues et incertitudes, il est apparu essentiel de prévoir un encadrement strict, minimisant la prise de risque pour les personnes, quand bien même celles-ci se prêtent volontairement à la recherche sans en attendre de bénéfice individuel direct. Définies pour la première fois par la loi de 1988 relative à la protection des personnes qui se prêtent à des recherches biomédicales 86 ( * ) , les règles applicables ont été régulièrement actualisées tout en reposant sur l'idée constamment réaffirmée que l'intérêt des volontaires qui se prêtent à une recherche clinique doit toujours primer sur l'intérêt de la science et de la société, conformément aux principes éthiques fondamentaux consacrés dès 1964 dans la déclaration d'Helsinki 87 ( * ) .
La loi de 1988, dite « Huriet-Sérusclat » , a ainsi introduit le principe d'une protection obligatoire des patients fondé un devoir d'information des volontaires, la nécessité de recueillir leur consentement écrit et l'obligation pour le promoteur d'une recherche clinique de souscrire un contrat d'assurance spécifique pour couvrir les risques encourus.
Afin d'évaluer le respect de ces principes et les protocoles de recherche, la loi institue dans chaque région un comité consultatif de protection des personnes qui se prêtent à la recherche biomédicale (CCPPRB). Pour chaque projet, elle impose l'identification d'un promoteur - personne morale (laboratoire pharmaceutique, prestataire de service, association, établissement de soins ou de recherche) ou physique -, qui prend l'initiative de l'essai clinique, et d'un investigateur principal, médecin chargé de diriger et de surveiller la réalisation de l'essai. La recherche fait l'objet d'une contractualisation entre le promoteur et le ou les centres d'investigation.
Ce régime a été renforcé par la loi de 2004 relative à la politique de santé publique 88 ( * ) qui permet l'application en France de la directive européenne du 4 avril 2001 89 ( * ) harmonisant les règles de vigilance des essais thérapeutiques entre les Etats membres. Cette loi confère à l'Agence française de sécurité sanitaire des produits de santé (Afssaps) - ancêtre de l'ANSM - la mission de veiller à ce que les conditions d'un consentement éclairé soient réunies ainsi que la possibilité de mener des investigations en cours d'essai par l'intermédiaire d'un comité d'éthique indépendant. Elle a transformé les CCPPRB en comités de protection des personnes (CPP) et rendu leur avis non plus consultatif mais décisionnel.
Le cadre législatif aujourd'hui applicable aux essais cliniques (articles L. 1121-1 à L. 1126-12 du code de la santé publique) est issu de la loi relative aux recherches impliquant la personne humaine de 2012, dite loi « Jardé » 90 ( * ) , qui n'est devenue applicable qu'en 2016 par l'édiction d'une ordonnance prise sur le fondement de la loi de modernisation de notre système de santé du 26 janvier 2016 91 ( * ) , dans l'attente par le Gouvernement de la révision des règles européennes applicables en la matière 92 ( * ) . Le décret d'application de la loi « Jardé » 93 ( * ) précise les modalités de réalisation des recherches impliquant la personne humaine (RIPH) et ajuste les règles relatives au rôle et au fonctionnement des CPP et de l'ANSM. Elle distingue plusieurs catégories de recherche en fonction des niveaux de risque, parmi lesquelles celles requérant le plus haut niveau de vigilance sont dénommées « recherches interventionnelles qui comportent une intervention non justifiée par sa prise en charge habituelle » (1° de l'article L. 1121-1 du code de la santé publique).
Pour ces dernières, le démarrage d'un essai clinique est soumis à la double condition d'un avis favorable d'un CPP et d'une autorisation de l'ANSM (art. L. 1121-4). Le promoteur a le choix d'introduire, simultanément ou non, la demande d'avis et celle d'autorisation.
En vertu de l'article L. 1121-11 du code de la santé publique, les volontaires, recensés sur un fichier national, font l'objet d'un examen médical préliminaire et aucune contrepartie financière directe ou indirecte hormis un remboursement des frais exposés ne peut leur être versée. Cette indemnisation est limitée à 4 500 euros pour une période de douze mois consécutifs. Une période d'exclusion entre les recherches doit être respectée. Enfin, les personnes bénéficiant d'une protection renforcée (mineurs, femmes enceintes, majeurs sous tutelle, etc.) ne peuvent être inclus dans les essais sauf en cas de bénéfice majeur direct possible pour leur santé.
Le CPP, dont la mission principale est de garantir la protection des volontaires, rend son avis sur les conditions de validité de la recherche au regard principalement des critères suivants 94 ( * ) :
- la protection des personnes, au premier rang desquelles les participants ;
- l'adéquation, l'exhaustivité et l'intelligibilité des informations écrites à fournir ainsi que la procédure à suivre pour obtenir le consentement éclairé, et la justification de la recherche sur des personnes incapables de donner leur consentement éclairé ;
- la nécessité éventuelle d'un délai de réflexion ;
- la nécessité éventuelle de prévoir, dans le protocole, une interdiction de participer simultanément à une autre recherche ou une période d'exclusion ;
- la pertinence de la recherche, le caractère satisfaisant de l'évaluation des bénéfices et des risques attendus et le bien-fondé des conclusions ;
- l'adéquation entre les objectifs poursuivis et les moyens mis en oeuvre ;
- la qualification du ou des investigateurs ;
- les montants et les modalités d'indemnisation des participants ;
- et enfin les modalités de recrutement des participants.
Le promoteur a l'obligation de soumettre à l'ANSM un protocole de recherche, qui se fonde sur une revue de la littérature scientifique et démontre la nécessité de réaliser une nouvelle recherche. En application de l'article L. 1123-12 du code de la santé publique, l'agence évalue l'objectif, les conditions de réalisation et de déroulement de l'essai, les modalités d'inclusion, d'information, de traitement et de surveillance des volontaires par les médecins investigateurs ainsi que les procédures de recueil des informations sur l'efficacité et la tolérance des médicaments. Une fois l'autorisation octroyée et l'essai débuté, l'ANSM est tenue informée des effets indésirables graves et inattendus pouvant être liés au médicament expérimental et de tout fait nouveau lié à la recherche susceptible de remettre en cause la sécurité des personnes se prêtant à la recherche. Le cas échéant, elle peut prendre toute décision de suspension ou d'interdiction. L'ANSM inspecte en outre certains sites d'essais cliniques.
Par ailleurs, en vertu des bonnes pratiques cliniques (BPC), dont la dernière version date du 24 novembre 2006, toute recherche doit faire l'objet de contrôles de qualité réalisés en début et en cours d'essai sous la responsabilité du promoteur de la recherche. Cette mission échoit en général aux assistants de recherche clinique (ARC) du promoteur.
En mars et juin 2016, à la suite de l'accident survenu à Rennes au cours d'un essai de phase I conduit par la société Biotrial pour le compte du laboratoire Bial chez un volontaire sain avec première administration chez l'homme d'une nouvelle molécule, ces règles de vigilance ont été encore renforcées. En particulier, le promoteur d'un essai doit désormais informer sans délai l'ANSM, le CPP et l'ARS de tout fait nouveau survenant dans le cas d'un essai de première administration ou utilisation d'un médicament expérimental chez des volontaires sains, en suspendre l'administration ou l'utilisation et prendre toutes les mesures de sécurité appropriées dans l'attente de l'adoption de mesures définitives.
2. Les essais cliniques comme voie d'accès précoce à des médicaments innovants
Dans certaines situations, pour les patients qui présentent une résistance aux traitements classiques ou qui demeurent sans solution thérapeutique efficace, des essais de phase précoce peuvent permettre d'accélérer la mise à disposition de nouvelles stratégies thérapeutiques . Ces essais, qui regroupent des caractéristiques d'essais de phases I et II, avec une évaluation concomitante de la sécurité du dosage et de l'efficacité, permettent d'accéder à un nouveau médicament plusieurs années avant sa commercialisation, de 3 à 4 ans selon le Leem.
Ce type d'essai remet en cause la distinction traditionnelle entre la recherche et le soin, alliant les deux approches dans une même démarche. Il soulève de nouveaux enjeux éthiques et juridiques, au regard notamment des conditions d'accès aux soins et de sélection des volontaires. La grande majorité des essais précoces sont aujourd'hui mis en oeuvre en cancérologie où les molécules à l'essai visent très souvent des cibles protéiques anormales caractéristiques d'un type particulier de tumeur dans le cadre d'une médecine de précision. Si ce type d'essais est en plein essor, il ne concerne ainsi qu'un petit nombre de malades. Comme l'indiquent Valérie Gateau et Philippe Amiel, « en cancérologie, les essais précoces sont les premiers tests d'administration d'une molécule, ou combinaison de molécules, chez l'être humain, à la suite des essais précliniques en laboratoires et/ou sur l'animal. Ils concernent environ 2500 malades par an, soit moins de 1 % de ceux qui participent à des essais cliniques en France tous les ans. Ces essais de phase I non randomisés visent à évaluer la dose maximale tolérée. Ils sont conduits sur un petit nombre de patients (de moins d'une dizaine à une trentaine). Contrairement aux essais dans d'autres pathologies, en raison de la toxicité des molécules testées en cancérologie, il n'y a que peu d'études menées sur des volontaires sains. » 95 ( * )
Les autorités ont progressivement pris la mesure de l'importance des essais précoces pour développer l'innovation thérapeutique et des initiatives structurantes ont vu le jour, en particulier pour permettre un accès plus égalitaire des personnes malades . C'est l'un des objectifs poursuivis dans le cadre des plans Cancer avec la mise en place à compter de 2010 des centres d'essais cliniques de phase précoce (Clip) labellisés par l'Institut national du cancer (INCa). Actuellement, 16 Clip existent, dont 6 en pédiatrie, qui sont labellisés jusqu'en 2019. Une attention particulière a ainsi été portée à l'amélioration de la couverture territoriale et au développement d'une activité de phase précoce en cancérologie pédiatrique.
Ainsi que l'a indiqué l'INCa, ce réseau vise tout autant à améliorer l'accès à l'innovation qu'à augmenter la visibilité et l'attractivité internationales des centres français de recherche. La moitié de l'activité de phase I des Clip est réalisée par le département d'innovation thérapeutique et d'essais précoces (DITEP) de l'Institut Gustave Roussy que vos rapporteurs ont pu visiter au cours d'un déplacement.
|
Le département d'innovation thérapeutique
A travers son département d'innovations thérapeutiques et d'essais précoces (DITEP), l'Institut Gustave Roussy a fait des essais précoces un moteur de l'innovation thérapeutique en cancérologie. Son approche combinée des soins et de la recherche, à travers la réalisation d'essais qui répondant de façon concomitante aux objectifs poursuivis au cours des phases I et II, vise à mettre en évidence l'efficacité d'une molécule de manière accélérée. Le délai d'accès aux innovations peut ainsi être ramené à 4-6 ans au lieu de 8 environ . Selon le DITEP, « à l'issue d'un essai clinique de phase I, la maladie régresse ou se stabilise chez près de 50 % des patients . » 96 ( * ) Le DITEP a été créé en 2013 à partir du service des innovations thérapeutiques et des essais précoces, première unité d'hospitalisation française intégralement dédiée aux essais précoces en cancérologie mise en place en 2008. Sa transformation en un département à part entière se fondait sur la volonté d'atteindre une masse critique, de fluidifier les processus et d'optimiser les performances. Aujourd'hui, le DITEP est le plus grand centre d'essais de phase I en France et en Europe et le septième dans le monde. En 2016, 417 recherches biomédicales ont été mises en oeuvre, dont 366 essais thérapeutiques. En 2017, 3 617 patients ont été inclus dans 438 recherches biomédicales, soit 28 % des 13 109 patients traités pour un cancer. L'Institut Gustave Roussy est le promoteur de la moitié des études menées par le DITEP. Le nombre annuel d'essais précoces en cours a connu une hausse constante, passant de 41 en 2010 à 104 en 2017. Les principales difficultés identifiées par les équipes de très haut niveau rencontrées par vos rapporteurs tiennent aux délais d'approbation réglementaire des essais précoces : pour l'année 2017, sur 25 essais précoces, le délai d'autorisation par l'ANSM s'élève à 121 jours en moyenne (la fourchette étant de 32 à 212 jours), et le délai d'avis pour les CPP à 91 jours (de 43 à 234). Le DITEP regrette en outre l'impossibilité pour les coordonnateurs académiques d'interagir avec les autorités réglementaires dans un esprit constructif pour faciliter la mise en place des essais. S'agissant enfin des CPP, ils relèvent l'expertise très variable de leurs membres et l'absence d'harmonisation des pratiques. Au total, une inquiétude existe quant à la défiance croissance des promoteurs industriels à développer leur activité en France. |
B. LA NECESSITÉ D'ACCÉLÉRER L'ACCÈS DES PATIENTS AUX ESSAIS CLINIQUES DANS DES CONDITIONS OPTIMALES DE SÉCURITÉ
1. L'attractivité de la France pour la recherche clinique est discutée, en particulier pour les essais de phase précoce
a) Un positionnement français stable dans une Europe qui décroît
Dans un contexte de compétition internationale fortement accrue au cours des dernières années, l'attractivité de la France pour la recherche clinique est devenue une préoccupation de tout premier plan, qui suscite des réactions mitigées, parfois contradictoires. Si la qualité de la recherche scientifique et clinique française est unanimement reconnue, force est de constater un sentiment général de décrochage . Selon les indications fournies à vos rapporteurs, il semble, de manière particulièrement illustrative, qu'au niveau mondial les discussions des autorités françaises avec les représentants de l'industrie pharmaceutique se focalisent en premier lieu sur la question de l'accès aux essais cliniques dans notre pays, avant même de porter sur celle de la fixation des prix des médicaments bénéficiant d'une AMM. La perception de l'environnement français de la recherche clinique est ainsi empreinte d'une certaine prudence. Pointant la diminution du nombre d'essais de phase précoce et la place décroissante de la France dans les études cliniques réalisées en Europe, la plupart des industriels appellent en effet de leurs voeux la mise en place de procédures plus rapides et faciles pour l'autorisation des essais.
Vos rapporteurs regrettent l'insuffisance des données de source institutionnelle permettant d'objectiver cette situation par un état des lieux consolidé de la recherche clinique tant académique qu'industrielle . Parmi les différentes études disponibles, celle réalisée périodiquement par le Leem dresse néanmoins un portrait nuancé des essais cliniques à promotion industrielle et ses conclusions convergent avec les constats établis devant vos rapporteurs au cours des auditions 97 ( * ) . Le Leem alerte sur « certains clignotants (qui) passent à l'orange » s'agissant de la longueur des délais de mise en place des essais et du recul de la France dans les études cliniques réalisées en Europe, notamment face à la concurrence de pays de l'Est.
De fait, si la France continue d'appartenir aux grands acteurs de la recherche clinique mondiale, sa situation est surtout celle d'une stagnation, voire d'un non-rattrapage dans une Europe dont la place décroît face à la montée en puissance de pays qui présentent un cadre réglementaire moins contraignant ou de moindres exigences au plan des garanties éthiques , à l'instar des États-Unis, lesquels concentrent 60 % des essais cliniques réalisés dans le monde, ou de la Chine.
Ce constat d'une Europe en perte de vitesse est bien identifié par les institutions européennes qui y ont vu une incitation à modifier le cadre juridique défini à cet échelon. Dans une évaluation de la directive sur les essais cliniques de 2001, aujourd'hui abrogée, la Commission européenne dressait en effet le triple constat d'une forte chute du nombre d'essais cliniques entre 2007 et 2010 (de l'ordre de 25 %), d'une hausse importante de leurs coûts et d'un allongement substantiel des délais de mise en place. Elle relevait que la directive « est critiquée par toutes les parties concernées (des patients aux entreprises du secteur, en passant par les chercheurs) au motif qu'elle a provoqué dans l'Union européenne un net désintérêt pour la recherche axée sur le patient et les études connexes. De fait, le nombre de demandes d'autorisation portant sur des essais cliniques dans l'Union européenne a baissé de 5 028 à 4 400 entre 2007 et 2010. Cette tendance entraîne une forte érosion de la compétitivité de l'Europe dans le secteur de la recherche clinique et a donc une incidence négative sur la mise au point de traitements et de médicaments nouveaux et innovants. » 98 ( * )
b) Un retard pour les essais de phase précoce
Selon les informations communiquées à vos rapporteurs, en 2016, l'ANSM a délivré 756 autorisations d'essais cliniques de médicaments, soit un nombre inférieur à ceux enregistrés les deux années précédentes. Cette baisse serait toutefois le reflet d'une nouvelle organisation au sein de l'agence. L'année 2017 enregistre une nouvelle baisse, avec 727 essais autorisés pour les médicaments . De manière constante, un tiers des promoteurs sont académiques et deux tiers industriels.
Autorisation des essais cliniques en faveur des médicaments
|
2012 |
2013 |
2014 |
2015 |
2016 |
2017 |
|
|
Nombre d'autorisations délivrées |
705 |
899 |
821 |
928 |
756 |
727 |
Source : ANSM, rapport d'activité pour 2017 (20/09/2017), et réponses au questionnaire de vos rapporteurs
Toutes phases confondues, environ 55 % des essais sont déposés en oncologie. Cette discipline concentre à elle seule environ 90 % des études de phase I, ce qui reflète l'importance croissante prise par cette discipline dans les développements et la vague d'innovations actuels.
Répartition des essais cliniques autorisés par domaine thérapeutique 99 ( * )
|
2013 |
2014 |
2015 |
|
|
Oncologie, hématologie, immunologie
|
379 |
345 |
426 |
|
Cardiologie, endocrinologie, gynécologie
|
110 |
85 |
224 |
|
Neurologie, psychiatrie, antalgie, rhumatologie, pneumologie, ORL, ophtalmologie, stupéfiants |
262 |
217 |
542 |
|
Anti-infectieux, hépato-gastroentérologie, dermatologie, maladies rares |
125 |
149 |
229 |
|
Vaccins et produits biologiques |
24 |
25 |
68 |
Source : ANSM, rapport d'activité pour 2015 (29/12/2016)
Malgré leur demande réitérée en ce sens auprès des autorités concernées, vos rapporteurs ne se sont pas vu communiquer de données statistiques suffisamment détaillées pour acquérir une vision d'ensemble de l'évolution des essais cliniques réalisés en France au cours du temps, en fonction des phases et des domaines d'intervention, des durées de mise en oeuvre, du nombre de volontaires concernés ainsi que des résultats obtenus.
Il apparaît néanmoins clairement que la position de la France pour les essais de phase précoce demeure insatisfaisante alors que leur nombre est en constante augmentation au niveau mondial. Le Leem estime que si la France se caractérise par le dynamisme de la recherche clinique en oncohématologie, elle fait face une baisse globale des études de phase précoce qui représentent 17 % des études en 2016 contre 20 % en 2014. Cet affaissement de la position française dans les essais de phase I serait en partie lié à l'affaire Biotrial, qui aurait engendré une dégradation de la perception de l'environnement français. En outre, certaines aires thérapeutiques apparaissent délaissées comme par exemple la neurologie et la psychiatrie qui ne représentent toutes les deux que 2 % des études de phase I contre 11 % en 2014, et toutes phases confondues, 6 % en 2016 contre 10 % en 2014 100 ( * ) . Pour les phases II et III, le Leem souligne que la France subit la concurrence d'autres pays qui ont une faculté d'inclusion beaucoup plus grande. Le ministère des solidarités et de la santé relève pour sa part que « si pour les essais de phase I, la France ne parvient pas à rattraper son retard, déjà ancien, sur les pays européens les plus compétitifs, elle demeure en tête pour le nombre d'essais de phase III réalisés en 2017 (204 essais cliniques contre 182 en Allemagne). Toutes catégories confondues, la France appartiendrait au trio de tête constitué du Royaume-Uni et de l'Allemagne ». 101 ( * )
Vos rapporteurs sont convaincus que la France dispose d'atouts indéniables, avec des scientifiques de très haut niveau, des établissements d'excellence et des prises en charge de grande qualité. Ces avantages comparatifs ne doivent néanmoins pas conduire à occulter les critiques qui se font entendre sur une perte de vitesse de la France , sur les inégalités territoriales liées à la centralisation des essais principalement dans les grandes villes et sur la rareté des essais cliniques dans certains domaines comme par exemple la pédiatrie. Il convient en particulier d'éviter l'effet de cliquet que peut engendrer la réalisation d'un essai de phase I dans un autre pays dans la mesure où toute délocalisation risque d'entraîner celle des essais de phases suivantes. Enfin, compte tenu des spécificités de leur modèle économique et de leur spécialisation poussée, un enjeu particulier existe pour les biotechs, dont les besoins de financement sont importants dès le stade le plus précoce de la recherche.
2. Plusieurs freins sont à lever pour accélérer la mise en place des essais cliniques et la mise à disposition de médicaments innovants aux volontaires
De l'avis général, la longueur des procédures administratives d'autorisation des essais cliniques et leur caractère inadapté aux enjeux de l'innovation constituent ainsi l'un des principaux freins au renforcement de l'attractivité du territoire français. Au-delà des questions de financement de la recherche, qui dépassent le cadre du présent rapport, réunir les conditions d'une recherche clinique réactive et dynamique implique tout d'abord des adaptations organisationnelles , tant des CPP que de l'ANSM, en particulier au regard des nouvelles règles européennes qui trouveront prochainement à s'appliquer.
Si les autorités n'ont pas su, en tout cas pour le moment, se doter d'indicateurs robustes de suivi des délais d'autorisation, en particulier pour les avis rendus par les CPP, chacun s'accorde sur le fait que ces délais sont très fréquemment dépassés . Selon le Leem, le délai médian entre la soumission du dossier de recherche clinique à l'ANSM et son autorisation est passé de 55 jours en 2014 à 57 jours en 2016. Le délai médian pour les avis rendus par les CPP demeure de 62 jours comme en 2014. Au total selon le Leem, la France « reste encore pénalisée par la longueur des délais avant le démarrage de l'essai. Faute de moyens accordés à l'ANSM comme aux CPP, les délais administratifs risquent encore de s'allonger, d'autant plus que l'ANSM se voit confier, aux termes de la loi Jardé, de nouvelles tâches d'évaluation méthodologique. De plus, la désignation des CPP par tirage au sort tel que prévu par la loi Jardé suscite les plus grandes réserves : en effet, ce tirage au sort implique que tous les CPP soient en mesure d'expertiser l'ensemble des protocoles de recherche dans toutes les pathologies. Or, l'enquête 2016 montre que 50 % des études cliniques sont évaluées par 9 des 40 CPP. »
a) De nouvelles exigences à respecter en application du nouveau règlement européen
Publié le 27 mai 2014, le règlement européen relatif aux essais cliniques de médicaments à usage humain 102 ( * ) a pour ambition d'accroître l'attractivité et les capacités de l'Union européenne pour la recherche biomédicale, en partant du constat d'une perte de vitesse de l'Europe à une époque de fort développement des innovations, en particulier dans le domaine de la génomique.
Il réforme en profondeur les conditions d'autorisation des essais cliniques dans l'Union européen en prévoyant une double évolution :
- d'une part, il renforce le rôle centralisateur de l'agence européenne du médicament (EMA) dans le sens d'une évaluation coordonnée des demandes d'autorisation d'essais cliniques et de leurs modifications. Dès lors qu'un essai est conduit dans au moins un État membre, la demande d'autorisation par le promoteur devra être déposée sur un portail européen unique . Celui-ci a vocation à regrouper l'ensemble des informations relatives aux essais et sera le support de tous les échanges entre les promoteurs, les autorités administratives des Etats membres participant à un essai clinique ainsi que les comités d'éthique de ces pays. Certaines données seront rendues accessibles au public ;
- d'autre part, le règlement prévoit une instruction des demandes en deux étapes : la première dans le cadre d'une évaluation coordonnée entre Etats membres concernés aboutissant à une conclusion unique (partie I), la seconde dans le cadre d'une évaluation par chaque État membre conduisant à une conclusion nationale (partie II). La partie I, assurée par un État membre rapporteur, vise à évaluer la demande au plan scientifique. La partie II concerne l'évaluation au plan éthique. Chaque État membre formule ensuite une décision nationale qui intègre les conclusions des parties I et II. Le règlement européen précise ainsi les rôles respectifs des autorités nationales compétentes en matière d'évaluation des essais cliniques de médicaments et des comités d'éthique.
Le règlement, publié au Journal Officiel de l'Union européenne le 27 mai 2014, s'appliquera dès la mise à disposition du portail européen, qui devrait intervenir courant 2019. Il implique une évaluation concomitante d'une demande d'essai clinique sur un médicament par l'ANSM et le CPP, débouchant sur une notification unique, par l'ANSM au promoteur, de la décision de l'agence et de l'avis du comité.
Pour une demande initiale, le délai au terme duquel l'ANSM et le CPP doivent s'être prononcé sur la validité d'un essai clinique est ainsi fixé à 45 jours et 91 jours en cas de questions complémentaires posées au promoteur. Une disposition d'« accord tacite » est prévue, selon laquelle en l'absence de réponse dans le délai imparti, l'essai est considéré comme accepté (le silence vaut acceptation).
|
Le règlement européen de 2014 sur les
essais cliniques :
Le règlement européen relatif aux essais cliniques de médicaments à usage humain impose aux États membres des échéances calendaires très resserrées qui doivent être respectées, faute de quoi l'autorisation d'un essai clinique est réputée acquise. La demande du promoteur doit être validée par l'État membre rapporteur dans un délai de dix jours à compter du dépôt du dossier de demande. L'État membre rapporteur transmet, par l'intermédiaire du portail de l'Union, la partie I finale du rapport d'évaluation, y compris sa conclusion, au promoteur et aux autres États membres concernés, dans un délai de quarante-cinq jours à compter de la date de validation. Chaque État membre concerne conclut son évaluation dans un délai de quarante-cinq jours à compter de la date de validation et soumet, par l'intermédiaire du portail de l'Union, la partie II du rapport d'évaluation, y compris la conclusion, au promoteur. Ce délai peut être prolongé de trente et un jours au maximum pour obtenir des informations complémentaires de la part du promoteur. Chaque État membre concerné fait savoir au promoteur si l'essai clinique est autorisé, s'il l'est sous conditions ou si l'autorisation est rejetée. La notification est effectuée sous la forme d'une décision unique dans un délai de cinq jours à compter de la date de rapport ou du dernier jour de l'évaluation (partie II). Si l'État membre concerné n'a pas notifié sa décision au promoteur dans les délais prévus, la conclusion sur la partie I du rapport d'évaluation est réputée être celle de l'État membre concerné s'agissant de la réponse à la demande d'autorisation de l'essai clinique. L'article 4 du règlement dispose que les États membres « veillent à ce que les délais et les procédures pour l'examen par les comités d'éthique soient compatibles avec les délais et procédures établis dans le présent règlement en ce qui concerne l'évaluation de la demande d'autorisation d'un essai clinique ». |
En France, l'application du règlement européen est assurée par l'ordonnance précitée du 16 juin 2016 103 ( * ) qui modifie le code de la santé publique dans sa rédaction issue de la loi « Jardé », par l'insertion d'un chapitre dédié constitué de l'article L. 1124 1. L'article 4 de l'ordonnance prévoit que l'évaluation scientifique et technique des essais cliniques sera réalisée par l'ANSM, les CPP se menant une évaluation au strict plan éthique.
La bonne application de ces règles nécessite une adaptation des procédures et des acteurs au plan national, en particulier un renforcement des relations entre l'ANSM et les CPP. Des évolutions ont été engagées en ce sens, qui devront être approfondies, en particulier pour renforcer la capacité d'intervention des CPP.
b) Renforcer les CPP et leur expertise
Au nombre de 39, les CPP sont désignés par les ARS et agréés par le ministère chargé de la santé pour une durée de 6 ans. Chacun est doté de la personnalité juridique et reçoit une dotation de l'État destiné à couvrir ses frais de fonctionnement.
|
La composition des comités de protection des personnes (CPP) La composition des CPP est établie de façon à « garantir leur indépendance et la diversité des compétences dans le domaine de la recherche impliquant la personne humaine et à l'égard des questions éthiques, sociales, psychologiques et juridiques » (article L. 1123-1 du code de la santé publique). Chaque CPP se compose ainsi de 14 membres, nommés par le représentant de l'État dans la région, qui se divisent en deux collèges paritaires : - un 1 er collège scientifique composé de 4 personnes qualifiées en recherche biomédicale dont au moins 2 médecins et 1 biostatisticien ou épidémiologiste, 1 médecin généraliste, 1 pharmacien hospitalier et 1 infirmier ; - un 2 e collège sociétal composé de 7 personnes qualifiées : 1 en matière d'éthique, 1 psychologue, 1 travailleur social, 2 juristes ainsi que 2 représentants d'associations de patients ou d'usagers du système de soins. Ces membres interviennent à titre bénévole et sont tenus à une obligation d'indépendance vis-à-vis des investigateurs et des promoteurs. |
En application de la loi « Jardé », les CPP sont chargés d'émettre un avis sur l'ensemble des projets de recherche impliquant la personne humaine (RIPH), quel que soit l'objet de l'essai clinique. Une commission nationale des recherches impliquant la personne humaine (CNRIPH) a pour mission d'assurer leur coordination (article L. 1123-1-1 du CSP). Conformément à la volonté du législateur, les comités ont perdu leur compétence régionale et sont devenus compétents pour l'ensemble du territoire, les dossiers de recherche étant répartis de manière aléatoire entre les CPP par une procédure de tirage au sort effectué par le secrétariat de la CNRIPH.
L'attention de vos rapporteurs a été attirée sur les difficultés, bien identifiées, auxquelles font face les CPP dans la réalisation de leur mission, malgré tout l'engagement et le dévouement de leurs membres dont il faut rappeler qu'ils sont bénévoles. Le niveau d'expertise et de réactivité des 39 CPP apparaît en effet insuffisant ou, à tout le moins, inégal. Ces difficultés sont principalement de trois ordres :
- un manque d'expertise pour les essais particulièrement complexes, en particulier ceux spécifiques aux cancers , dont les modalités sont devenues extrêmement techniques. Plusieurs personnes auditionnées par vos rapporteurs ont notamment indiqué que de nombreuses questions posées par certains CPP aux promoteurs des essais témoignent fréquemment d'un manque d'expertise de leurs membres, avec le risque soit de retarder les procédures, soit, au contraire, de passer à côté d'un enjeu important ou d'une difficulté. Dans certains cas, la composition des CPP apparaît inadaptée au regard des compétences requises pour la compréhension des protocoles de recherche. A titre d'exemple, une grande partie des CPP ne seraient pas en mesure de satisfaire à l'obligation de recourir à un pédiatre pour les projets impliquant des mineurs de moins de 16 ans. ;
- une forte hétérogénéité des niveaux et des pratiques , qui traduit une harmonisation insuffisante des méthodes d'évaluation, aggravée par l'absence d'indicateurs de suivi ;
- et enfin des dysfonctionnements administratifs , souvent liés à un manque de moyens qui réduit la disponibilité de certains CPP, en particulier pendant certaines périodes de congé comme par exemple durant l'été.
Au total, l'intégralité des CPP n'est pas en capacité de travailler efficacement dans des délais raisonnables, souvent faute de spécialistes disponibles ou d'experts mobilisables, en particulier pour les essais de phase I. Les modalités de désignation des CPP fondées sur le principe du tirage au sort sont de nature à accroître ces difficultés.
Dans ces conditions, vos rapporteurs sont convaincus qu'il convient d'adapter le système du tirage au sort afin que celui-ci s'applique à un groupe restreint de CPP spécialisés en fonction du domaine de l'essai clinique. Ce dispositif va dans le sens de celui adopté en mai dernier à l'occasion de l'examen en première lecture à l'Assemblée nationale d'une proposition sur l'expertise des CPP 104 ( * ) . Partant du constat que les demandes ne peuvent être traitées dans le délai imparti de 45 jours, ce texte prévoit un dispositif qui vise à orienter la demande vers un comité doté de la compétence nécessaire à l'évaluation, que cette compétence soit présente en son sein ou disponible dans son réseau. Il est ainsi prévu que le tirage au sort soit effectué parmi les CPP disponibles et qui disposent de la compétence nécessaire. Il n'apparaît en effet pas opportun d'inclure dans le tirage au sort les comités qui ne sont pas en mesure de mobiliser cette compétence indispensable à l'évaluation de la démarche éthique du projet.
Vos rapporteurs jugent en outre indispensable de renforcer le niveau d'expertise des CPP, notamment par la mise en place de formations adaptées et l'accès à des experts rapidement mobilisables. A cet égard, il serait opportun d'accroître les interactions avec l'ANSM dans le sens d'une mutualisation de l'expertise.
L'harmonisation des procédures d'évaluation doit également se poursuivre sous l'égide de la CNRIPH.
Enfin, il apparaît indispensable de renforcer les moyens administratifs dont disposent les comités pour leur permettre de faire face à leurs missions et de mieux valoriser le travail des bénévoles.
|
Proposition n° 16 : Renforcer les comités de protection des personnes (CPP) et leur expertise : - Adapter le système du tirage au sort en prévoyant que celui-ci s'applique à un groupe restreint de CPP spécialisés en fonction du domaine concerné par l'essai clinique ; - Renforcer le niveau d'expertise des CPP, notamment par la mise en place de formations adaptées et la mise à disposition d'experts en cas de besoin ; - Poursuivre l'harmonisation des procédures d'évaluation ; - Renforcer les moyens administratifs dont disposent les comités. |
Sous la pression du nouveau cadre juridique européen, de l'attribution de missions supplémentaires et d'un contexte budgétaire contraint, l'ANSM fournit d'importants efforts pour adapter ses procédures internes et prioriser ses missions à partir de l'idée qu'il convient d'adapter l'évaluation en fonction des risques.
Un groupe de travail du « comité d'interface sur les essais cliniques » a notamment engagé à l'automne 2017 une réflexion sur les conditions à réunir pour renforcer l'attractivité de la France dans le domaine de la recherche clinique tout en garantissant la sécurité des patients.
Pour répondre à l'enjeu spécifique de l'instruction des essais cliniques de phases précoces, l'ANSM a mis en place, fin 2017, une cellule dédiée au sein de sa direction des politiques d'autorisation et d'innovation (DPAI) 105 ( * ) . Ses ressources sont issues à la fois d'autres directions et de recrutements externes. Aujourd'hui, malgré des moyens que vos rapporteurs jugent encore relativement limités, les représentants de l'agence estiment que les compétences nécessaires pour instruire les demandes d'essais précoces dans des délais raisonnables sont réunies 106 ( * ) .
Afin d'anticiper l'application du nouveau règlement européen relatif aux essais cliniques de médicaments à usage humain, l'agence est par ailleurs entrée dès septembre 2015 dans une phase pilote associant à titre expérimental les promoteurs académiques et industriels qui le souhaitent ainsi que les CPP. Consécutivement à l'entrée en vigueur de la loi « Jardé », les 39 CPP ont été intégrés au dispositif. Une démarche a également été engagée pour mieux coordonner l'activité de l'agence avec celle des CPP. Un nouveau système d'information est en cours de conception pour faciliter les échanges entre les promoteurs et les comités ainsi qu'entre ceux-ci et l'ANSM.
Dans ses évaluations périodiques de la phase pilote, l'agence souligne que celle-ci a permis la mise en place d'interactions constructives entre les parties prenantes (promoteurs, CPP et ANSM), une mobilisation importante des CPP ainsi qu'une forte participation des promoteurs académiques et industriels. L'avancée de l'instruction du dossier est devenue plus visible, avec la mise à disposition d'un seul calendrier d'instruction des demandes et une facilitation des démarches liée à l'envoi du dossier complet le même jour et à la réception d'une notification unique.
Aux yeux de vos rapporteurs, le bilan de cette phase pilote est relativement encourageant. L'ANSM demeure cependant sous très forte tension et, dans la période récente, le délai moyen dans lequel les avis sont rendus connaît une augmentation.
Selon l'agence, au 28 septembre 2017, deux ans après le début de cette phase expérimentale, 260 dossiers ont été déposés et 210 gérés par la cellule dédiée, soit 14 % des dossiers déposés à l'agence. Près de la moitié des dossiers gérés ont concerné des médicaments en oncologie et en hématologie. Le nombre de promoteurs académiques ayant participé s'élève à 33 (comptant pour 97 dossiers déposés) et celui des promoteurs industriels à 40 (113 dossiers déposés). Sur les 193 demandes clôturées, 127 ont abouti à une autorisation de l'ANSM et un avis favorable du CPP concerné avec un délai moyen d'instruction de 68,9 jours .
Evolution du délai moyen de notification finale
|
Demandes clôturées |
Nbre autorisation
|
Délai moyen |
|
|
Bilan 6 mois (au 28/03/16) |
26 |
21 |
57,4 j |
|
Bilan 12 mois (au 30/09/16) |
89 |
73 |
64,3 j |
|
Bilan 18 mois (au 31/05/17) |
128 |
98 |
65,5 j |
|
Bilan 24 mois (au 28/09/17) |
193 |
127 |
68,9 j |
Source : ANSM (Direction de l'évaluation, 17 octobre 2017)
Dans ces conditions, vos rapporteurs ne peuvent qu'appeler de leurs voeux la poursuite des adaptations organisationnelles . Au-delà de l'optimisation des procédures, qui doit constituer un axe d'amélioration permanent, la question des moyens de l'agence pour faire face à ses missions doit demeurer un point de vigilance. A cet égard, l'agence estime avoir « mis en place une organisation au regard des moyens alloués visant à permettre un traitement optimisé des essais cliniques dans le cadre règlementaire actuel. Les moyens futurs devront être adaptés en lien avec la stratégie que la France veut mener au sein d'un cadre européen et le positionnement qu'elle souhaite occuper. » 107 ( * ) Dans la perspective de l'application prochaine du règlement européen, en particulier de la disposition d'autorisation tacite, vos rapporteurs estiment que les enjeux justifient un renforcement des moyens de l'ANSM dédiés à l'instruction.
|
Proposition n° 17 : Augmenter les moyens de l'ANSM dédiés à l'instruction des essais cliniques précoces et poursuivre l'optimisation de la procédure de gestion des essais cliniques en adaptant l'évaluation en fonction des risques. |
c) Simplifier la convention unique
La mise en oeuvre, dans un établissement de santé, des recherches biomédicales à vocation industrielle mobilisant les compétences et les moyens de cet établissement, les coûts et surcoûts générés pour celui-ci sont facturés à l'industriel promoteur de la recherche en tant que prestation de service. Compte tenu de l'enjeu représenté par les délais de mise en place des recherches du point de vue de la compétitivité internationale, la nécessité de réduire le délai de signature des conventions qui lient l'industriel promoteur de la recherche à l'établissement de santé dans lequel se déroule l'investigation a été prise en compte dans le contrat stratégique de filière « Industries et Technologies de Santé », signé le 5 juillet 2013 par le gouvernement et les organisations professionnelles représentant les industries de santé. En vertu de sa mesure 19, une convention unique remplace, depuis fin 2016, le contrat unique pour les études de recherche clinique à promotion industrielle.
Avant 2014, les laboratoires pharmaceutiques étaient obligés de négocier les coûts de leur recherche avec chaque établissement de santé partenaire. Il s'agissait d'une procédure complexe qui allongeait les délais de contractualisation. Selon le Leem, ces délais s'élevaient à 8 mois en moyenne mais pouvaient aller jusqu'à plus de deux ans et demi. La convention unique associe désormais, pour un même lieu de recherche, le promoteur industriel, l'établissement de santé et, le cas échéant, une structure tierce destinataire de contreparties financières. Elle a vocation à être utilisée à l'identique par tous les établissements de santé français participant à une même recherche biomédicale. Cette mesure de simplification administrative et de transparence a été étendue à l'ensemble des établissements de santé par la loi de modernisation de notre système de santé du 26 janvier 2016 (article L. 1121-16-1 du code de la santé publique) 108 ( * ) . La convention unique est pilotée par un établissement coordonnateur avec un délai de signature limité à 60 jours. Selon le Leem, en 2015, 331 études ont démarré avec la signature de 1 838 conventions et en 2016, 493 études avec 3 116 conventions.
Vos rapporteurs relèvent que l'utilité de la convention unique est appréciée diversement par les personnes auditionnées : si les interlocuteurs de la mission ont généralement souligné son caractère positif, il a été souligné qu'elle n'avait pas eu, pour tous, les effets escomptés . UniCancer estime en particulier que « la convention unique, qui était destinée à réduire les délais, a dans le réseau des CLCC produit l'effet inverse, avec un allongement des délais et une perte significative de financement des surcoûts par les industriels. »
Vos rapporteurs estiment que la mise en place de la convention unique a constitué une importante mesure de rationalisation des procédures. Les efforts de simplification doivent néanmoins se poursuivre et la convention unique être appropriée par l'ensemble des établissements investigateurs. Il s'agit d'ailleurs de l'un des objectifs assignés dans le cadre du CSIS, ces nouvelles modalités de contractualisation entre investigateurs et promoteurs n'apportant pas encore entière satisfaction.
|
Proposition n° 18 : Poursuivre la simplification de la convention unique et la généraliser à l'ensemble des établissements. |
C.
« L'UTILISATION TESTIMONIALE ECLAIRÉE
ET SURVEILLÉE » :
UN DISPOSITIF QUI SOULÈVE
DES ENJEUX D'ÉTHIQUE
ET DE SECURITÉ
Au regard des délais de mise à disposition d'un médicament innovant aux patients sans solution ou en échec thérapeutique, la question de l'accès le plus précoce possible à une molécule prometteuse, y compris en phase préclinique, est posée.
C'est le sens d'un amendement adopté au Sénat à l'occasion de l'examen en première lecture du projet de loi de financement de la sécurité sociale pour 2018 109 ( * ) , à l'initiative de notre collègue René-Paul Savary, visant à instituer une « utilisation testimoniale éclairée et surveillée » (UTES) d'un médicament en développement.
Il s'agirait de rendre accessibles à des patients, incurables et volontaires, des molécules susceptibles de déboucher sur une innovation thérapeutique, même si ces substances sont à un stade d'évaluation clinique précoce et dès lors que la preuve de l'existence d'éléments scientifiques, pré-cliniques ou cliniques, démontrant leur potentiel intérêt thérapeutique chez l'homme et permettant de présumer de leur sécurité, pourrait être fournie. L'UTES constituerait ainsi un nouveau mode d'accès au médicament, à côté de l'essai clinique, de l'AMM et de l'ATU.
Dans sa version adoptée au Sénat, le recours à ce dispositif serait conditionné au consentement éclairé du patient et se fonderait sur une décision médicale. Sa mise en oeuvre s'effectuerait sous la surveillance de l'ANSM. Selon ses concepteurs, ce dispositif aurait vocation à s'appliquer notamment dans le domaine des maladies neuro-dégénératives pour faire accéder des patients à des traitements expérimentaux dans un cadre dérogeant à la législation sur les essais cliniques. Le recours à l'UTES dans ce domaine se justifierait, d'après les auteurs de l'amendement, « non seulement en ce que les essais cliniques y sont les plus longs, à cause de l'évolution très lente des neuro-dégénérescences, mais également parce que les médications neuro-protectrices présentent par nature un profil de sécurité encourageant. »
Pour vos rapporteurs, le délai de mise en place des essais cliniques pouvant, incontestablement, constituer un frein en cas d'urgence thérapeutique, l'intérêt d'un dispositif tel que l'UTES est une question majeure.
La plupart des personnes auditionnées par vos rapporteurs, en particulier les associations de patients, ont toutefois exprimé des réserves dans la mesure où ce dispositif soulève un fort enjeu de sécurité et d'éthique : son recours doit se concevoir avec une certaine vigilance car il conduit à mettre à la disposition des patients des médicaments très peu évalués et aux effets secondaires méconnus ou sous-évalués. Comme le souligne la HAS, une grande incertitude règne à cette étape particulièrement précoce du développement, ce qui implique, compte tenu des risques encourus, une approche éthique de l'accord que le patient donnerait à son utilisation, même si cet accord était parfaitement éclairé. De son côté, France Assos Santé « s'oppose à la déclinaison en France d'un tel dispositif... qui, en répondant à des demandes individuelles, remettrait en cause les principes collectifs de notre système d'évaluation du bénéfice-risque, d'accès aux traitements et de recueil des données » 110 ( * ) .
Il apparaît que dans certaines situations, des dispositifs juridiques proches de ceux existants, en particulier un dispositif d'ATU précoce et réactive comme celui présenté à la proposition n°1 du présent rapport, pourraient atteindre l'objectif poursuivi par l'UTES.
L'ANSM estime pour sa part que « les essais cliniques et les ATU peuvent répondre à cette problématique et sont déjà des dispositifs d'accès aux traitements innovants. Si un tel dispositif concernant l'utilisation testimoniale des innovations pharmaceutiques pourrait être mis en oeuvre, il conviendrait que ce dispositif soit particulièrement encadré afin de ne pas être en concurrence avec l'encadrement des essais cliniques. » En tout de cause, ainsi que l'a indiqué l'INSERM, « si la question est celle des rares AMM obtenues en phase I de façon exceptionnelle pour des médicaments en cancérologie ou pour les maladies rares par exemple, il convient effectivement d'encadrer ces usages avec un suivi en vie réelle du médicament ». A cet égard, il pourrait être envisagé, dans un premier temps, d'évaluer l'UTES dans un cadre expérimental en se fondant sur les réflexions en cours dans d'autres pays et les expériences acquises par exemple aux États-Unis, où un dispositif analogue a récemment été adopté dans le cadre du « Right to try act » - à savoir un « droit à essayer » - qui s'adresse aux patients en fin de vie sans solution thérapeutique 111 ( * ) .
La mise en place d'une expérimentation permettrait par ailleurs d'alimenter la réflexion sur des questions importantes soulevées par l'UTES, relatives à la prise en charge financière des traitements expérimentaux, à la volonté des laboratoires de les mettre à disposition et aux responsabilités juridiques encourues par les acteurs concernés. Compte tenu de l'ensemble de ces considérations, vos rapporteurs estiment que la réflexion sur l'UTES doit se poursuivre et sa valeur ajoutée s'apprécier au regard des améliorations apportées aux dispositifs d'accès précoce déjà existants. Selon les indications qui leur ont été communiquées, les services du ministère des solidarités et de la santé et l'ANSM seraient d'ailleurs saisis d'une commande en ce sens, visant à analyser l'état des besoins.
EXAMEN EN COMMISSION
___________
Réunie le mercredi 13 juin 2018 sous la présidence de M. Alain Milon, président, la commission examine le rapport d'information, fait au nom de la mission d'évaluation et de contrôle de la sécurité sociale, de M. Yves Daudigny, Mmes Catherine Deroche et Véronique Guillotin sur l'accès précoce à l'innovation dans le domaine des produits de santé.
M. Alain Milon , président . - Nous passons à l'examen du rapport de nos collègues Yves Daudigny, Catherine Deroche et Véronique Guillotin sur l'accès précoce à l'innovation en matière de produits de santé.
Il s'agit d'un travail réalisé dans le cadre du programme de travail annuel de la mission d'évaluation et de contrôle de la sécurité sociale, la Mecss, dont je salue le président, Jean-Noël Cardoux.
Je rappelle que la publication des rapports de la Mecss doit être autorisée par la commission dans son ensemble.
Notre commission s'est déjà penchée, il y a deux ans, sur la question du prix du médicament, ce qui nous avait donné l'occasion d'esquisser cette question de l'innovation qui soulève autant d'espoirs pour les patients et les équipes soignantes que de défis pour notre système de santé et, au-delà, pour notre protection sociale.
Mme Catherine Deroche , co-rapporteure . - Dans le cadre de la Mecss, nous avons souhaité approfondir les conditions de l'accès des patients aux thérapies innovantes, sous l'angle des délais. Nous partions du sentiment diffus selon lequel ces délais seraient devenus en France plus longs que dans d'autres pays.
Je remercie le président de la Mecss, Jean-Noël Cardoux, de nous avoir confié cette mission sur un sujet complexe mais à forts enjeux : nos auditions ont confirmé combien il était identifié comme crucial, tant par les laboratoires que les pouvoirs publics, les patients ou encore et surtout les professionnels de santé. Je remercie d'ailleurs ceux d'entre vous qui se sont montrés assidus à nos travaux.
Le contexte est en effet inédit : l'accélération des innovations, notamment en oncologie avec l'arrivée des thérapies ciblées ou de l'immunothérapie, offre des promesses formidables ; parallèlement, le coût de ces traitements, réel ou fantasmé, représente un défi pour la soutenabilité de notre système de santé.
Derrière des discours nuancés, le constat général est que la France dispose de sérieux atouts, par son expertise en matière d'essais cliniques et le dispositif des autorisations temporaires d'utilisation (ATU), tête de proue d'une politique ambitieuse d'accès précoce à l'innovation. Toutefois, un certain nombre de freins existent, que nous avons cherché à identifier et que nos propositions visent à lever.
Nous avons ciblé notre réflexion sur le médicament, en excluant le dispositif médical qui relève de procédures différentes. Toutefois, la dynamique de ce secteur est telle qu'il mériterait à lui seul une prochaine mission !
Notre champ d'étude est déjà vaste. Nous allons l'aborder étape par étape, des essais cliniques à la commercialisation des médicaments après leur autorisation de mise sur le marché (AMM), en passant par le dispositif spécifique des ATU qui constituait le point de départ de notre réflexion. Nous vous avons remis un schéma qui retrace les grandes étapes de cette chaîne.
M. Yves Daudigny , co-rapporteur . - J'en commence par les autorisations temporaires d'utilisation (ATU).
Nous avons pu constater, au cours de nos auditions, un attachement aussi unanime qu'enthousiaste de l'ensemble des acteurs à ce dispositif, ce qui est assez rare pour être souligné. Le mécanisme a en effet permis, depuis plus de vingt ans, de mettre la quasi-totalité des médicaments innovants à la disposition des malades atteints de pathologies graves, souvent mortelles, parfois plus d'un an avant la délivrance de l'AMM - ce qui représente bien sûr un gain de chances considérable.
Cette mise à disposition à la fois précoce et universelle est fondée sur un socle de principes toujours d'actualité. Mis en place en 1994 dans l'objectif d'assurer un accès précoce à ce qui constituait alors les nouveaux médicaments anti-VIH, le dispositif des ATU a été conçu comme un mécanisme temporaire et dérogatoire, visant à répondre aux situations d'impasse thérapeutique pour les patients atteints de pathologies graves ou rares et dont l'état de santé ne permet pas d'attendre davantage.
Il existe deux types distincts d'ATU : les ATU nominatives, délivrées pour un patient nommément désigné à la demande de son médecin, et les ATU de cohorte, qui s'adressent à des groupes de patients traités et surveillés en application d'un protocole, et qui sont délivrées à la demande des industriels. Le dispositif a été conçu comme universel : il permet l'accès de tous les patients concernés en tout point du territoire.
Tout médicament faisant l'objet d'une ATU est pris en charge par l'assurance maladie dès l'octroi de cette autorisation, à prix libre - donc fixé par les laboratoires - (appelé « indemnité ») et sans évaluation préalable par la Haute Autorité de santé. Ces modalités de financement dérogatoire contribuent bien évidemment à garantir l'accès rapide des patients français à l'innovation. Elles constituent également pour les industriels, selon les représentants du Leem (organisation professionnelles des entreprises du médicament), « un élément différenciant positif pour l'attractivité de la France ».
En dépit de la permanence de ces principes structurants, le dispositif des ATU a profondément changé de nature depuis sa création. Il ne s'agit plus, ou plus seulement, d'un mécanisme compassionnel, visant à prendre en compte le profil thérapeutique particulier de certains patients, mais d'un dispositif structuré d'accès précoce au marché destiné à de grands volumes de patients. Un peu plus de 27 000 ATU nominatives ont ainsi été délivrées en 2016, portant sur 205 médicaments ; les ATU de cohorte bénéficient quant à elles à plus de 10 000 patients chaque année.
Mme Véronique Guillotin , co-rapporteure . - Cette évolution a été largement provoquée par le retour de l'innovation médicamenteuse. En particulier, deux chocs sur les dépenses ont résulté de l'arrivée dans le dispositif de nouvelles molécules très innovantes : en 2014 et 2015 tout d'abord, avec l'arrivée des nouveaux traitements contre l'hépatite C ; en 2016 ensuite, avec l'entrée de nouveaux traitements d'immunothérapie contre le cancer, dont les anti-PD1. Les dépenses d'ATU sont ainsi très concentrées sur quelques médicaments : en 2015, ces anti-PD1 et les médicaments destinés à traiter l'hépatite C ont représenté 75 % de la dépense totale des médicaments sous ATU.
En conséquence, alors que la dépense liée aux ATU plafonnait à 110 millions d'euros annuels jusqu'en 2013, elle a atteint deux pics successifs à un milliard d'euros en 2014 et 2016.
Ce mouvement devrait connaître une forte intensification au cours des prochaines années, notamment avec l'arrivée prochaine dans le dispositif d'une nouvelle génération d'immunothérapie, les Car-T cells, dont le coût devrait être sans rapport aucun avec celui des médicaments que nous connaissons aujourd'hui.
Cette évolution pose problème à plusieurs titres.
En premier lieu, le fonctionnement du dispositif semble aujourd'hui rencontrer ses limites scientifiques. Je m'explique : les ATU sont délivrées pour un médicament, dans une ou plusieurs indications données, en amont de la première autorisation de mise sur le marché (AMM) pour ce produit. Il en résulte que, passée la délivrance de l'AMM, le périmètre de l'ATU se fige. En d'autres termes, toute extension d'indication devient ainsi impossible.
Or, cette réglementation apparaît inadaptée au mode d'action des médicaments les plus innovants contre le cancer que constituent les immunothérapies. Dans la mesure où ils visent à renforcer le système immunitaire du patient, ils peuvent être efficaces contre plusieurs types de cancers différents. On a ainsi pu observer, lors de l'utilisation des anti-PD1 que j'évoquais plus tôt, le développement rapide d'indications parallèles ou successives.
Dans les conditions actuelles, le mécanisme ne garantit pas aux patients de recevoir le traitement le plus efficace disponible.
A notamment été cité, au cours de nos auditions, le cas du nivolumab, qui, après avoir bénéficié d'une ATU en 2014, est aujourd'hui autorisé et admis au remboursement contre les mélanomes et les cancers du poumon. Il est cependant impossible de le prescrire à des patients atteints, notamment, de cancers de la vessie ou ORL, alors même que les essais cliniques sont positifs et que le produit est déjà sur le marché aux États-Unis dans ces indications. Il nous paraît dès lors indispensable d'autoriser au plus vite les extensions d'indication pour les médicaments bénéficiant ou ayant bénéficié d'une ATU.
M. Yves Daudigny , co-rapporteur . - En second lieu, la forte croissance des dépenses a nécessité la mise en place d'un nouveau mécanisme de régulation des montants versés au titre des ATU, qui cristallise aujourd'hui un très fort mécontentement chez les industriels.
Il me faut ici être un peu technique. Dans tous les cas où le prix du médicament finalement déterminé après l'AMM est inférieur à l'indemnité fixée par le laboratoire au cours de la période d'ATU, la différence est reversée à l'assurance maladie par le laboratoire sous la forme d'une remise. L'article 97 de la LFSS pour 2017 a prévu que ce montant est désormais calculé par rapport à un prix de référence basé sur une prévision des volumes de vente pour les trois années suivant la sortie du dispositif ATU.
Ce mode de calcul est unanimement dénoncé par les laboratoires comme remettant en cause la lisibilité et la prévisibilité du mécanisme de sortie des ATU. En particulier, le montant de la remise est difficile à absorber par les petites entreprises de biotechnologies, qui constituent aujourd'hui des acteurs importants de l'innovation médicamenteuse.
Du fait de la particulière complexité de ce dispositif, il n'apparaît pas raisonnable de poursuivre son application plus longtemps. L'objectif de maîtrise des dépenses de l'assurance maladie doit être concilié avec la nécessaire attractivité du dispositif d'ATU, afin de continuer à garantir cet accès essentiel à l'innovation médicamenteuse pour les patients français. Certains laboratoires ont laissé entendre qu'ils pourraient envisager de ne plus solliciter d'ATU ; nous regrettons ce qui s'apparente à une menace qui pèserait in fine sur les patients. Toutefois, ce constat n'est pas encore confirmé par les statistiques.
Nous estimons par ailleurs que la maîtrise des dépenses d'ATU constitue une impérieuse nécessité, dans le but de continuer à garantir aux patients français un accès non rationné à l'ensemble des innovations. Il n'apparaît pas soutenable de mettre à la charge de l'assurance maladie, pendant une durée parfois très longue, des indemnités fixées à un niveau élevé par les laboratoires.
Les ATU intervenant aux étapes précoces du développement d'un médicament, alors que la phase de recherche est parfois toujours en cours, il n'est pas possible de disposer à ce stade de données cliniques exhaustives et fiables ; les ATU concernent donc des médicaments présumés très innovants. Or, les évaluations conduites par la Haute Autorité de santé après l'obtention de l'AMM montrent que tous les médicaments sous ATU ne constituent pas nécessairement des ruptures d'innovation ce qui influe sur le prix ensuite fixé. Cette situation n'est satisfaisante ni pour les industriels, qui se voient alors contraints de rembourser des montants importants à l'assurance maladie, ni pour les pouvoirs publics, qui versent des indemnités élevées ne correspondant pas toujours au service thérapeutique rendu aux patients.
Pour répondre à ces deux difficultés, nous estimons qu'il pourrait être envisagé de rendre le dispositif des ATU plus rapide et plus souple, mais révisable à tout moment sur la base de données d'utilisation obligatoirement produites au cours des phases d'ATU et de post-ATU. Une telle solution permettrait de payer l'innovation au plus juste prix, de bénéficier de données de suivi précieuses, et, pour les laboratoires, d'accéder à une valorisation commerciale plus rapide et plus sécurisée de leurs produits.
Mme Catherine Deroche , co-rapporteure . - Les difficultés relatives aux ATU ne se comprennent que dans le cadre plus général des procédures de mise sur le marché « de droit commun » du médicament.
Après l'AMM, délivrée au niveau européen pour les médicaments innovants, s'engage au plan national une procédure essentielle puisqu'elle détermine la prise en charge des médicaments et la fixation de leur prix. Elle comprend deux principales étapes : tout d'abord, l'évaluation par la HAS du service médical rendu (SMR) et du progrès thérapeutique, l'amélioration du service médical rendu (ASMR) ; puis, sur la base des résultats de cette évaluation, la négociation et la fixation du prix devant le Comité économique des produits de santé (CEPS).
Cette procédure est séquencée, alors qu'en Allemagne - modèle qui nous a été fréquemment cité - le médicament est mis à disposition dès l'AMM, sur la base d'un prix librement fixé par les laboratoires (comme pour nos ATU) ; l'évaluation se déroule en parallèle et le prix définitif est fixé au terme de la première année de commercialisation.
La procédure doit se dérouler en théorie en 180 jours. Toutefois, ce délai serait de près de 300 jours en 2016 pour les médicaments non générique en ville d'après les données du CEPS. Selon un indicateur suivi par l'EFPIA, la fédération européenne des laboratoires, dont le périmètre est différent, le délai moyen entre l'AMM et la commercialisation serait de plus de 500 jours en France, en augmentation et dans une position très moyenne en Europe loin derrière l'Allemagne, le Royaume-Uni ou encore l'Italie. Ces données sont à prendre avec précaution car elles n'intègrent pas les produits sous ATU ce qui dégrade la position de la France. On peut d'ailleurs regretter qu'un indicateur de délai ne soit pas suivi de manière rigoureuse au niveau du ministère.
Néanmoins, ces chiffres révèlent des freins. Quels sont-ils ?
En ce qui concerne l'évaluation, son caractère rigoureux est reconnu. Pour autant, ses modalités sont réinterrogées : pour la Haute Autorité de santé (HAS), l'octroi d'AMM plus précoces par l'agence européenne du médicament, sur la base de données considérées comme immatures, rend sa tâche plus complexe ; pour les laboratoires et notamment les biotechs, en résultent des évaluations de l'ASMR (c'est-à-dire du progrès thérapeutique) jugées parfois timides, qui pèsent dans la fixation du prix. Les critères de l'évaluation sont aussi perçus illisibles et imprévisibles.
Les tensions se concentrent sur la question du prix : d'un côté les exigences des laboratoires sont jugées aberrantes, notamment au regard des promesses non tenues de certains médicaments ; de l'autre des industriels considèrent que l'innovation n'est pas reconnue à sa juste valeur, avec une pression sur les prix plus forte en France que dans plusieurs pays voisins. Les prix nets de remises n'étant pas publics, il nous a été toutefois difficile d'objectiver ce constat.
S'il ne faut pas trop noircir le tableau, la situation n'est pas satisfaisante, avec le sentiment général d'un système qui subit l'innovation et son coût et un déficit de confiance entre les acteurs.
Dans ce contexte, nos propositions visent à donner à la fois plus de visibilité et de souplesse, pour mieux adapter la grille de lecture à la réalité des innovations et des besoins.
Mme Véronique Guillotin , co-rapporteure . - D'une manière générale, au-delà du Conseil stratégique des industries de santé (le CSIS), qui se déroule tous les deux ans - cette année avec des conclusions attendues début juillet - un cadre d'échange plus pérenne entre les acteurs permettrait de développer une vision prospective qui fait encore défaut. Plusieurs pays ont mis en place des systèmes de ce type, ce qui permettrait notamment de mieux cibler les efforts sur les priorités en accélérant l'accès au marché des produits fléchés comme susceptibles de répondre à un besoin thérapeutique en apportant un progrès pour les malades.
En outre, nos outils et procédures sont sans doute à repenser pour concilier sécurité des patients, accès précoce et maîtrise budgétaire. Une évolution évoquée par la HAS comme par des industriels serait de rendre possible, pour des médicaments prometteurs mais insuffisamment développés au stade de leur autorisation de mise sur le marché, un remboursement temporaire, conditionné à l'apport de données supplémentaires dans un délai préétabli.
De même, et si en nous mesurons la complexité technique, il serait intéressant d'expérimenter, sur des périmètres d'abord restreints, des modes de fixation de prix plus souples (comme l'idée de prix différenciés par indication), ou plus évolutifs en fonction de l'efficacité vérifiée en vie réelle. Cela permettrait de sortir d'un système à certains égards trop rigide, alors que l'accélération des innovations exige aujourd'hui davantage de fluidité et de nouvelles réflexions sur le partage des risques avec les industriels.
Enfin, nous avons été sensibles au problème spécifique des maladies rares : les AMM demandées par les industriels portent sur des indications parfois très pointues. Dans des situations « de niche », peu rentables, des prescriptions hors-AMM sont souvent indispensables. L'Institut national du Cancer a évoqué l'idée d'encadrer ces prescriptions sur la base de référentiels de bon usage, pour sécuriser ces pratiques. Cette piste permettrait tout à la fois de renforcer la pertinence des prescriptions et de s'adapter à la réalité des besoins.
Concernant toujours les maladies rares, l'évaluation, en particulier médico-économique qui mériterait d'être de façon générale développée, doit mieux s'adapter, dans sa méthodologie, à leurs spécificités. Là aussi, le cadre actuel peut conduire à des situations d'incompréhension entre les acteurs.
M. Yves Daudigny , co-rapporteur . - L'équité d'accès aux traitements innovants et souvent onéreux est-elle par ailleurs encore assurée ?
À l'hôpital, la diffusion des traitements onéreux repose sur un outil essentiel, créé en parallèle de la tarification à l'activité : la « liste en sus », c'est-à-dire les médicaments pris en charge par l'assurance maladie en sus des prestations forfaitaires d'hospitalisation (les groupements homogènes de séjour ou GHS). 400 indications y figurent pour une dépense de 3 milliards d'euros en 2016, en hausse de 23 % depuis 2009 et concentrée sur une quinzaine de spécialités notamment en oncologie.
De l'avis quasi-général, le fonctionnement de cette liste n'est pas satisfaisant, en raison de son caractère trop sédimenté mais aussi de critères de gestion rigides et contestés. Pour les professionnels de santé, certains produits jugés prometteurs ne s'y retrouvent pas. C'est ainsi l'équité d'accès entre les patients qui est mise en cause, selon les territoires et le mode de prise en charge en ville ou à l'hôpital. La présidente de la HAS a reconnu que l'ASMR (à savoir l'amélioration du service médical rendu), qui est un critère médical, n'avait pas vocation à définir un mode de prise en charge.
Nous proposons que les critères d'inscription et de radiation sur la liste en sus soient revus mais aussi que sa gestion plus dynamique soit rendue possible par une meilleure anticipation des sorties. Un assouplissement de ces critères devrait s'accompagner d'une plus forte responsabilisation des prescripteurs ; la LFSS pour 2018 ouvre la voie à une expérimentation sur ce champ, ce qui serait bienvenu.
Notre attention a également été appelée sur les enjeux du RIHN, le référentiel des actes innovants hors nomenclature, qui concerne les actes de biologie ; c'est le cas des tests « compagnons », nécessaires à la mise en oeuvre des thérapies ciblées contre le cancer. Les enjeux de pertinence des soins et de qualité de vie des patients sont majeurs puisqu'ils permettent des désescalades thérapeutiques, par exemple éviter une chimiothérapie pour certains patients.
L'enveloppe annuelle de 380 millions d'euros ne permet pas de financer l'ensemble des actes pris en charge par les établissements de santé qui représentent le double de cette somme en 2017. Ce risque financier induit des pratiques différenciées entre établissements et constitue un frein à la diffusion des tests. Comme pour la liste en sus, une gestion plus dynamique du RIHN est indispensable pour lui permettre de jouer son rôle de diffusion des innovations. Cela passe selon nous par une accélération de l'inscription à la nomenclature des actes passés en « routine » et leur réévaluation régulière.
Mme Véronique Guillotin , co-rapporteure . - Il nous faut enfin consolider le rôle des essais cliniques dans l'accès précoce aux innovations, dans un contexte de compétition internationale fortement accrue. L'attractivité de la France pour la recherche clinique suscite aujourd'hui des réactions mitigées, voire contradictoires. Si la qualité de la recherche française est à juste titre reconnue, nous avons perçu un sentiment général de décrochage : la plupart des industriels pointent la diminution du nombre d'essais de phase précoce et la place décroissante de notre pays s'agissant du nombre d'études réalisées. De fait, les éléments qui nous ont été communiqués par le Gouvernement font surtout état d'une stagnation de la France dans une Europe qui décroît face à des concurrents comme les États-Unis ou la Chine.
De l'avis général des personnes auditionnées, la longueur des procédures d'autorisation des essais cliniques et leur caractère inadapté aux enjeux de l'innovation constituent l'un des principaux freins au renforcement de l'attractivité du territoire. Chacun appelle de ses voeux la poursuite des adaptations organisationnelles d'ores et déjà amorcées, en particulier au regard des règles européennes qui imposent de nouveaux délais d'évaluation par les comités de protection des personnes (CPP) et l'Agence nationale de sécurité du médicament (ANSM).
Le niveau d'expertise et de réactivité des 39 CPP, chargés de rendre un avis sur les conditions de validité de la recherche au regard de critères principalement éthiques, apparaît insuffisant. Tous ne sont pas en capacité de remplir efficacement leur mission, souvent faute de spécialistes disponibles ou d'experts mobilisables, en particulier pour les essais de phase I. Dans certains cas, en particulier dans le domaine des cancers, leur composition apparaît inadaptée au regard de la complexité croissante des essais mis en oeuvre. S'y ajoutent une hétérogénéité des pratiques ainsi que des dysfonctionnements administratifs. Les modalités de désignation des CPP, qui se fondent depuis 2012 sur le principe du tirage au sort, sont de nature à accroître ces difficultés.
Nous formulons ainsi plusieurs propositions pour adapter le système du tirage au sort afin que celui-ci s'applique à un groupe restreint de CPP spécialisés en fonction du domaine de l'essai clinique. Ce dispositif va dans le sens de celui adopté par nos collègues députés à l'occasion de l'examen en première lecture d'une proposition de loi d'initiative centriste. Nous jugeons en outre indispensable de renforcer le niveau d'expertise des CPP, notamment par la mise en place de formations adaptées et l'accès à des experts rapidement mobilisables. L'harmonisation des procédures d'évaluation doit également se poursuivre. Enfin, il apparaît indispensable de renforcer les moyens administratifs dont ils disposent pour leur permettre de faire face à leurs missions.
Mme Catherine Deroche , co-rapporteure . - L'ANSM fournit quant à elle, c'est incontestable, des efforts pour adapter ses procédures internes et prioriser ses missions, mais il faut demeurer vigilant sur la question des moyens. Afin d'anticiper l'application du nouveau règlement européen relatif aux essais cliniques de médicaments à usage humain, l'agence est entrée dans une phase pilote associant à titre expérimental les promoteurs académiques et industriels ainsi que les CPP. L'avancée de l'instruction du dossier est ainsi devenue plus visible pour les promoteurs, avec la mise à disposition d'un seul calendrier d'instruction des demandes et une facilitation des démarches. Le bilan est encourageant. L'agence demeure cependant sous forte tension et, dans la période récente, le délai moyen dans lequel les avis sont rendus connaît une légère augmentation. Dans ces conditions, l'optimisation des procédures doit se poursuivre et les enjeux nous paraissent justifier un renforcement des moyens dédiés à l'instruction.
Quelques mots pour finir sur la question du dispositif connu sous le nom d'« utilisation testimoniale éclairée et surveillée » (UTES) adopté au Sénat dans le projet de loi de financement de la sécurité sociale pour 2018, à l'initiative de notre collègue René-Paul Savary. Il s'agirait de rendre accessibles à des patients, incurables et volontaires, des molécules qui sembleraient avoir une forte chance de déboucher sur une innovation thérapeutique, même si ces substances sont à un stade d'évaluation clinique précoce, voire très précoce.
Au regard des délais de mise à disposition d'un médicament innovant aux patients sans solution ou en échec thérapeutique, la question de l'accès le plus précoce possible à une molécule prometteuse, y compris en phase préclinique, est en effet clairement posée.
De l'avis général des personnes que nous avons auditionnées, le recours à l'UTES ne saurait cependant se concevoir qu'avec la plus grande vigilance. La plupart d'entre elles, en particulier les associations de patients, ont exprimé des réserves, voire une opposition franche, dans la mesure où le dispositif soulève un fort enjeu de sécurité et d'éthique. L'UTES conduit en effet à mettre à la disposition des patients des médicaments très peu évalués et aux effets secondaires méconnus ou sous-évalués : comme le souligne la HAS, une grande incertitude règne à cette étape particulièrement précoce du développement, ce qui implique, compte tenu des risques encourus, une approche éthique de l'accord que le patient donnerait à son utilisation, même si cet accord était parfaitement éclairé.
Il nous semble que, dans l'attente de l'accès d'une molécule au marché du médicament, les ATU nominatives pourraient constituer un recours. Selon les informations qui nous ont été communiquées, la direction générale de la santé et l'ANSM seraient d'ailleurs saisis d'une commande visant à analyser l'état des besoins. En tout état de cause, la réflexion doit se poursuivre.
Telles sont nos principales observations et recommandations.
M. Alain Milon , président . - Ce rapport a l'immense qualité de mettre le doigt là où cela fait mal. C'est d'ailleurs notre rôle.
Je veux vous alerter sur le projet de loi de révision constitutionnelle et les articles concernant les projets de loi de finances (PLF) et de financement de la sécurité sociale (PLFSS). Il est prévu que la commission des affaires sociales ne soit plus impliquée dans l'examen du PLFSS, qui serait de la compétence exclusive de la commission des finances. J'entends demander à ce que nous soyons saisis pour avis de droit. En effet, je vois mal nos collègues de la commission des finances se prononcer sur les ATU ou les médicaments innovants.
Mme Florence Lassarade . - Ce rapport fait le point sur un sujet très complexe. J'attire votre attention sur les examens biologiques multiparamétriques qui permettent de déterminer les dosages précis des médicaments anti-cancéreux, en fonction du métabolisme du patient, afin d'éviter des effets toxiques néfastes. Ce sont a priori des tests peu onéreux, qui rendent un service considérable. Toutefois, nous n'avons pas les moyens de les rendre obligatoires.
M. Gérard Dériot . - Je félicite les rapporteurs. J'ai participé à de nombreuses auditions, et ce rapport retranscrit de manière claire et exhaustive toute la complexité du sujet. Ce rapport doit être diffusé et avoir une véritable portée.
Pour revenir sur les propos de notre président, je considérerais comme une régression parlementaire de ne plus permettre à la commission des affaires sociales d'examiner le PLFSS. Laisser ces sujets à la commission des finances serait catastrophique, car de tels rapports sont nécessaires pour éclairer la décision publique.
M. Michel Amiel . - Je dois dire mon admiration pour la clarté de ce rapport qui porte sur un sujet très complexe. En ce qui concerne les ATU, je ne comprends pas pourquoi nous ne pouvons répondre de manière simple aux situations d'extension d'indication.
L'exemple de l'anti-PD1 dans le traitement du cancer est criant. Si ce médicament, vu son mode d'action, peut être efficace pour le traitement de certains cancers non inclus initialement dans le champ de l'ATU, pourquoi ne fait-on pas confiance aux oncologues qui connaissent les maladies et travaillent avec ces molécules au quotidien ?
L'utilisation testimoniale éclairée et surveillée constitue, au-delà des questions de sécurité, un fort enjeu éthique que vous avez souligné. Il est regrettable que ce sujet ne soit pas abordé dans le cadre de la consultation citoyenne et des travaux en cours sur la révision des lois de bioéthique. Il faut porter notre réflexion sur notre conception de l'éthique à la française, qui peut être un frein à l'innovation, au-delà de la dimension économique et financière bien évidemment centrale dans ces questions.
Mme Martine Berthet . - Il était urgent de faire ce travail et ce rapport mérite d'être diffusé. Il y a une attente forte à la fois de la part des patients et des laboratoires.
Pouvez-vous revenir sur la notion de prix net de remise. S'agit-il du remboursement versé par les laboratoires à l'assurance maladie après fixation du prix ?
M. René-Paul Savary . - J'ai participé à de nombreuses auditions, au cours desquelles nous avons entendu des vérités et des contre-vérités : en effet, les intérêts des uns sont à l'opposé de ceux des autres.
La France a été à la pointe de l'innovation mais elle est aujourd'hui en phase de décrochage. J'exprime une certaine déception par rapport à des propositions que j'aurais souhaitées un peu plus ambitieuses. Les États-Unis viennent ainsi de voter le « Right To Try Act », à savoir un « droit d'essayer ». Il faut replacer le patient au centre du dispositif. Nous savons qu'il existe des molécules innovantes en matière de lutte contre les maladies neurodégénératives notamment. Or, on s'abrite derrière la responsabilité éthique, économique, financière, celles des médecins ou des ministres. En attendant, des milliers de patients meurent. Certains patients accepteraient de prendre ce risque. C'est l'objet de l'utilisation testimoniale éclairée et surveillée. Nous pourrions proposer une mise en oeuvre à titre expérimental, en mettant en garde les patients. Le ministère de la santé n'y est pas insensible. Il est dommage que le Sénat sur ce point ne soit pas plus provocateur, quitte à choquer quelques-uns, pour lancer le débat et aller de l'avant.
Mme Catherine Deroche , co-rapporteure . - Je ne vous cache pas la complexité du sujet. Tout le processus est très compliqué depuis l'essai préclinique jusqu'à la commercialisation. C'est un véritable parcours du combattant.
En ce qui concerne l'UTES, nous avons tenu à inscrire dans le rapport l'état de la réflexion et ce qui se fait aux États-Unis. Cette réflexion chemine. Les ATU nominatives peuvent être une solution.
L'amendement adopté au Sénat dans le cadre du PLFSS pour 2018 a ouvert le débat. La direction générale de la santé et l'ANSM, comme je l'ai indiqué, sont saisies du sujet pour réfléchir à une solution. Nous demandons que les travaux se poursuivent.
Les problématiques sont très variées. Pour les industriels, se pose d'abord la question de la fixation du prix. Certaines administrations sont ouvertes, d'autres estiment que les exigences des laboratoires sont excessives. Or, on est en présence d'innovations qui vont parfois concerner très peu de personnes. On ne peut pas appliquer à ces médicaments innovants - notamment lorsque l'on voit l'évolution des traitements - les mêmes logiques et les mêmes procédures qu'aux médicaments plus classiques.
Il faut, pour ces médicaments innovants, des procédures plus rapides et mieux adaptées qui permettent de trouver de nouveaux équilibres, d'être plus réactif et de fluidifier les mécanismes de régulation des prix. Nous avons entendu les attentes des laboratoires et celles des administrations. Nous sommes également allés à la rencontre des équipes du département des essais précoces à l'Institut Gustave Roussy. Cela nous a beaucoup éclairés. Tous nous disent que nous avons en France une recherche de qualité. D'ailleurs, le congrès mondial du cancer qui s'est tenu aux États-Unis la semaine dernière a montré le nombre important de publications françaises. Les essais pourraient se faire en France, mais il y a des blocages. Nous sommes en France les champions de la règlementation et de la complexité ! D'autres pays ont des approches plus pragmatiques. L'innovation aura un coût, certes. Mais il faut également prendre en compte les économies qu'elle permet de réaliser. Aujourd'hui, nous sommes bloqués dans une vision à court terme, avec la préoccupation de ne pas dépasser l'objectif de dépenses annuelles fixé par l'ONDAM.
Les tests évoqués par Florence Lassarade ne sont pas correctement pris en charge alors qu'ils sont essentiels. Les modes de financement sont complexes. Cela renvoie aux difficultés que nous évoquions à travers l'exemple du RHIN, le référentiel des actes innovants hors nomenclature. Certains établissements de santé ont les moyens de pratiquer ces tests, d'autres pas. Il y a donc aujourd'hui une inégalité d'accès à des tests qui permettraient d'indiquer qu'une chimiothérapie n'est pas nécessaire, car elle ne serait pas efficace, ou alors quelle quantité de produit administrer aux patients sans atteindre une dose toxique. Les chercheurs du centre hospitalier universitaire d'Angers ont travaillé sur ce sujet. Ils savent comment limiter la toxicité du 5-FU contenu dans les chimiothérapies, qui peut conduire à des décès. Or, aujourd'hui, on bloque sur les modalités de remboursement de ces tests. Il faut que l'administration parvienne à réfléchir sur le long terme, en tenant compte du retour sur investissement - sans parler des bénéfices humains pour les patients eux-mêmes.
M. Yves Daudigny . - Pour revenir sur les propos du président, ce n'est pas la première menace qui pèse sur notre commission en ce qui concerne le PLFSS. J'ai le souvenir d'avoir déjà contribué à limiter les attaques sur le rôle de la commission des affaires sociales. L'attaque est aujourd'hui d'autant plus sérieuse que le financement de la sécurité sociale se fait désormais par la contribution sociale généralisée.
La question des extensions d'indication dans le cadre des ATU est revenue à longueur d'auditions. Tout le monde considère le système des ATU formidable, mais celui-ci est victime de son succès. Nous sommes confrontés à l'explosion des dépenses. Dès lors, l'administration se concentre davantage sur les moyens de réguler le coût du dispositif, plutôt que de s'attacher à en étendre le champ. En outre, depuis 1994, le contexte médical a changé. Les ATU sont nées à une époque où la thérapie génique ou l'immunothérapie n'existaient pas. Nous proposons des solutions. Le conseil stratégique des industries de santé va également formuler des propositions en juillet. Cette difficulté est bien identifiée, mais les solutions auront un impact financier.
Le prix facial, aussi appelé « prix listé », correspond au tarif de remboursement des médicaments. Une négociation entre l'industriel et le CEPS, couverte par le secret des affaires, conduit par ailleurs à déterminer des remises conventionnelles. Ces dernières sont collectées via les URSSAF et abondent le fonds de financement pour l'innovation pharmaceutique. Le montant des remises n'est pas public. Toutefois, le rapport annuel du CEPS retrace leur montant global. J'ai le souvenir d'un interlocuteur nous indiquant que ces remises peuvent atteindre jusqu'à 80 % du prix facial d'un médicament.
Pourquoi le prix facial est-il alors maintenu ? ll reste une référence et un prix d'affichage pour les autres pays. Il existe ainsi un mécanisme de garantie de prix européen pour certains médicaments innovants qui permet un alignement des prix pratiqués dans un panel de pays (Allemagne, Espagne, France, Italie et Royaume-Uni). Mais, en France, ce qui compte pour l'assurance maladie est le prix net des remises versées par le laboratoire.
M. René-Paul Savary . - Ces remises vont aux URSSAF...
Mme Véronique Guillotin . - En effet, mais elles sont ensuite reversées au fonds pour le financement de l'innovation pharmaceutiques et reviennent donc dans le circuit de l'assurance maladie, comme les systèmes complexes français aiment le faire....
Deux sujets m'ont interpellée lors des auditions. D'abord, le manque de confiance entre les acteurs, à savoir entre les laboratoires et les pouvoirs publics, se manifeste dans les propos tenus par les uns et les autres. Pour caricaturer la situation, il semble que l'administration se soit attachée à mettre en place des verrous pour limiter les profits des laboratoires ; or, ces verrous sont aussi des freins à l'innovation.
Le problème majeur relatif aux ATU concerne les extensions d'indication. Ces blocages sont incompréhensibles et sont un réel frein au déploiement des traitements innovants qui nous arrivent en oncologie.
Mme Michelle Meunier . - J'ai participé à plusieurs auditions avec intérêt. N'étant ni médecin, ni technicienne de ces sujets, je les ai abordés à travers le prisme du patient et de sa famille. L'équité d'accès aux innovations, quel que soit le lieu où l'on est traité, est un enjeu majeur. Il est insupportable pour les patients et leurs familles de s'entendre refuser un traitement en raison de son coût prohibitif. Le problème est politique.
La recherche n'est pas nulle en France. Mais en raison du poids des procédures, d'une certaine conception du principe de précaution, les compétences de nos chercheurs s'exportent, vers les États-Unis, la Chine, mais aussi d'autres pays européens. Nous devons être capables de commercialiser des innovations trouvées et pensées en France.
Enfin, il ne faut pas perdre du vue que le patient doit rester au centre de toutes ces réflexions.
M. Michel Amiel . - En ce qui concerne les extensions d'indication dans le cadre des ATU, ne serait-il pas possible d'imaginer une négociation avec les industriels pour une diminution du prix, dans la mesure où la population cible du médicament est élargie ?
M. Yves Daudigny , co-rapporteur . - Pour l'instant, il y a un blocage fort sur toutes les questions relatives au prix du médicament.
M. Gérard Dériot . - La procédure d'autorisation de mise sur le marché a été mise en place pour protéger les patients. Depuis 60 ans, le nombre d'accidents liés à l'usage de médicaments nouveaux est très faible. Je pense notamment au thalidomide, qui provoquait des malformations sur les enfants lorsqu'il était pris par leurs mères pendant la grossesse. Ce médicament a été commercialisé dans de nombreux pays européens mais, fort heureusement, pas en France.
Pour des produits de haute technicité, peut-être est-on dans l'hyperprotection ? A un moment donné, il faut savoir prendre un risque. Mais dans un contexte de judiciarisation de notre société, qui porterait ce risque ?
M. René-Paul Savary . - Le malade !
M. Gérard Dériot . - Je voulais rappeler que la régulation du secteur pharmaceutique répond d'abord à un objectif sanitaire, et non mercantile.
Le rapport est adopté à l'unanimité.
LISTE DES PERSONNES ENTENDUES
___________
• Agence nationale de sécurité du médicament et des produits de santé (ANSM)
Lotfi Boudali , directeur à la direction des médicaments en oncologie, hématologie, transplantation, néphrologie, thérapie cellulaire, produits sanguins et radiopharmaceutiques
Carole Le Saulnier, directrice des affaires juridiques et réglementaires
Marc Martin, directeur adjoint de la direction des politiques d'autorisation et d'innovation
• Haute Autorité de santé (HAS)
Pr Dominique Le Guludec, présidente
Isabelle Adenot , présidente de la commission nationale d'évaluation des dispositifs médicaux et des technologies de santé (CNEDiMTS)
Pr Christian Thuillez , président de la commission de la Transparence (CT)
Dr Chantal Bélorgey , directrice de l'évaluation médicale, économique et de santé publique
• Association des groupes internationaux pour la pharmacie de recherche (Agipharm)
Pierre Claude Fumoleau , président de Abbvie France
David Setboun , président directeur général de Biogen France
Emmanuelle Quilès , présidente de Jassen France
Mickaël Halimi , manager affaires publiques Nextep
Jean Monin , président de Amgen France
• Comité économique des produits de santé (CEPS)
Maurice-Pierre Planel
,
président
Jean-Patrick Sales
,
vice-président
• Les entreprises du médicament (Leem)
Philippe Lamoureux , directeur général
Thomas Borel , directeur des affaires scientifiques
Frédéric Chassagnol , président du groupe de travail « accès précoce à l'innovation » et directeur accès au marché, Laboratoire Roche
Fanny de Belot , responsable des affaires publiques
Annaïk Lesbats , chargée de mission affaires publiques
Fabrice Meillier , directeur des affaires réglementaires
• France Assos Santé
Christophe Duguet , AFM-Téléthon
Jean-Pierre Thierry , conseiller médical de France Assos Santé
Caroline Izambert , Aides
• Fédération hospitalière de France (FHF)
Dr Dominique Goeury , praticien hospitalier, chargée de mission produits pharmaceutiques au pôle finances
• Fédération des établissements hospitaliers et d'aide à la personne (Fehap)
Dr Françoise Durandière , conseillère médicale
Julie Boissier-Laine , adjointe de direction, chargée de recherche, référence et recours
• Fédération de l'hospitalisation privée (FHP)
Michel Ballereau , délégué général
Anne Mallet , secrétaire nationale de l'UNHPC (Union Nationale Hospitalière Privée de Cancérologie)
Béatrice Noëllec , directrice des relations institutionnelles et de la veille sociétale
• Unicancer
Pascale Flamant , déléguée générale
Pr. Pierre Fumoleau , directeur de l'ensemble hospitalier de l'Institut Curie
• Institut national du cancer (INCa)
Pr Norbert Ifrah , président
Thierry Breton , directeur général
• Institut national de la santé et de la recherche médicale (Inserm)
Pr Yves Lévy , président directeur général
Claire Giry , directrice générale déléguée
• Direction générale de l'offre de soins (DGOS)
Dr Grégory Emery , adjoint à la sous-directrice du pilotage de la performance des acteurs d'offre de soins
• Direction générale de la santé (DGS)
Céline Perruchon , sous-directrice de la politique des produits de santé et de la qualité des pratiques et des soins
Nadine David , cheffe du bureau du médicament
• Direction de la sécurité sociale (DSS)
Mathilde Lignot-Leloup , directrice
Edouard Hatton , chef du bureau des produits de santé
• Caisse nationale d'assurance maladie (Cnam)
Nicolas Revel , directeur général
Michèle Surroca , responsable du département des produits de santé
• Pierre Besançon , ancien professeur d'université, investisseur en biotechs
• Collectif d'entreprises de biotechnologies dans les maladies rares en France
Dr Brigitte Calles , directeur affaires publiques, Vertex Pharmaceuticals
Dr Antoine Ferry , président CTRS
M. Armel de Gouvello , directeur accès au marché, Intercept Pharmaceuticals
• Coordonnateurs du 8ème Conseil stratégique des industries de santé (CSIS)
Vincent Lidsky , inspecteur général des finances
Noël Renaudin , ancien président du Comité économique des produits de santé
• Médecins du monde
Olivier Maguet , responsable de la campagne « Prix du médicament et systèmes de santé »
• Pr Jean-Paul Vernant , chef du service d'hématologie à la Pitié-Salpêtrière
• Pr Pascal Joly , coordinateur de centre expert et investigateur de projet de recherche au CHU de Rouen
• Dr Brigitte Roy-Geffroy , directeur exécutif de la Société française de dermatologie
• Pr Alain Astier , professeur en pharmacie clinique oncologique, chef de service de la pharmacie à l'hôpital Henri Mondor
• Pr Christophe Le Tourneau , responsable Department of drug development and innovation (D3i), Institut Curie
• Pr Jean-Marc Tréluyer , pharmacologie et recherche clinique Paris Descartes Cochin Necker
• Nicolas Labrune , conseiller au cabinet de Mme Agnès Buzyn, Ministre des solidarités et de la santé
• Celgene France
Christophe Durand , président directeur général
Yannick Sabatin , directrice des relations extérieures et de l'accès au marché
Ont par ailleurs transmis des contributions écrites :
• Roche
• Bristol-Myers Squibb France
• MSD France
LISTE DES DÉPLACEMENTS
___________
Jeudi 5 avril 2018
_____
• Institut Gustave Roussy - Département d'innovation thérapeutique et des essais Précoces (DITEP)
Pr Gilles Vassal , directeur de la recherche clinique (DRC)
Dr Christophe Massard , chef du département d'innovation thérapeutique et des essais précoces (DITEP)
Dr Aurélien Marabelle , directeur clinique du programme d'immunothérapie
Dr Eric Angevin, alliance manager (DITEP et direction de la recherche clinique (DRC))
Pr Jean-Yves Scoazec , chef du département biologie-pathologie, anatomopathologiste
Dr Etienne Rouleau , généticien
Grégory Vial , directeur de la stratégie et du développement
* 1 Créé en 2004, le CSIS est présenté comme « le lieu où s'élabore une vision stratégique du secteur des industries de santé commune aux pouvoirs publics et aux industriels et où sont proposées des réponses partagées. » (communiqué de presse du Premier ministre, 20 octobre 2017).
* 2 Le champ du présent rapport est ciblé sur le médicament, à l'exclusion du dispositif médical qui relève de procédures distinctes et mériterait de faire l'objet d'une étude à part entière.
* 3 Cf. encadré p. 23.
* 4 Cf. partie II.
* 5 Décret n° 94-568 du 8 juillet 1994 relatif aux autorisations temporaires d'utilisation de certains médicaments à usage humain et modifiant le code de la santé publique.
* 6 Loi n° 92-1279 du 8 décembre 1992 modifiant le livre V du code de la santé publique et relative à la pharmacie et au médicament.
* 7 Ce délai est prévu par le décret n° 2017-707 du 2 mai 2017 relatif à la valeur maximale du délai de dépôt d'une demande d'autorisation de mise sur le marché faisant suite à une autorisation temporaire d'utilisation d'une ou plusieurs indications d'un médicament.
* 8 L'examen du dossier par l'ANSM repose sur plusieurs critères : la sécurité d'emploi et l'efficacité du médicament dans l'indication revendiquée, le projet de protocole d'utilisation thérapeutique, le projet de notice d'information, les conditions de prescription et de délivrance, etc.
* 9 Rapport d'activité de l'ANSM pour l'année 2016.
* 10 Ce chiffre ainsi que ceux qui suivent sont donnés en excluant les patients traités par la spécialité Nalscue. Pour ce produit, l'ATUc consiste en la mise à disposition de naloxone (Nalscue) sous forme de spray nasal auprès des usagers de drogues pour le traitement d'urgence des surdoses aux opioïdes.
* 11 Lancé en avril 2014, le Promising Innovative Medicine (PIM) permet un accès à des médicaments avant leur autorisation de mise sur le marché. En novembre 2015, 11 médicaments avaient obtenu la qualification de PIM, majoritairement en oncologie (d'après l'Institut national du cancer, étude internationale sur l'innovation médicamenteuse en cancérologie, janvier 2018).
* 12 Voir notamment le rapport de l'OCDE « New Health Technologies. Managing Access, Value and Sustainability » de 2017, qui fournit des données comparatives sur l'accès précoces aux médicaments dans plusieurs États. Ce rapport, qui précise que des programmes d'accès précoces ont été introduits dans plusieurs pays de l'organisation, cite notamment le système des ATU françaises, mais aussi les dispositifs existant au Royaume-Uni et aux États-Unis.
* 13 Cette répartition est moins nette si l'on se réfère à l'ensemble du champ des ATU, qui inclut également les ATU nominatives. Selon les comptes de la sécurité sociale, 16 % des produits ayant bénéficié de ce statut en 2016 concernaient la neurologie, 15 % l'infectiologie et la parasitologie, 10 % les maladies cardiovasculaires et 7% la cancérologie.
* 14 Les éléments qui suivent sont tirés du Livre blanc du cercle de réflexion en immuno-oncologie (Crio), « Les défis de l'immunothérapie en oncologie. Réussir l'intégration de l'innovation en immunothérapie anticancéreuse dans la prise en charge du cancer en France », 2017.
* 15 Le principe d'action de ces anticorps est de ne plus freiner l'action anti-tumorale des cellules immunitaires. Le Livre blanc du Crio précité l'exprime en ces termes : « Les cellules tumorales développent à leur surface des molécules appelées ligand PD-L1. Ces molécules se lient avec des récepteurs appelés PD-1 situés à la surface des cellules immunitaires. En faisant cela, elles désactivent la vigilance des cellules immunitaires et peuvent ainsi se multiplier sans risque de destruction. Les traitements d'immunothérapie visent à empêcher cette interaction en introduisant des anticorps monoclonaux dans l'organisme du patient. Ces anticorps se lient de façon prioritaire soit aux récepteurs PD-1 (anti-PD1) soit aux ligands PD-L1 (anti-PDL1), ce qui réactive la vigilance des cellules immunitaires ».
* 16 Selon un article du Monde en date du 4 juin 2018, le principe de la thérapie par Car-T cells est le suivant : « des globules blancs, les lymphocytes T du patient, soldats de l'immunité, sont prélevés, cultivés in vitro, puis modifiés génétiquement de manière à leur faire exprimer un récepteur artificiel (le « CAR »), qui reconnaît spécifiquement les cellules de la tumeur à combattre. Après un délai de plusieurs semaines, ils sont ensuite réinjectés au patient, prêts à tuer ces cellules ».
* 17 Le rapport à la commission des comptes de la sécurité sociale de septembre 2017 rappelle que « la principale molécule innovante dans le traitement contre l'hépatite C, le sofosbuvir, commercialisé sous le nom Sovaldi®, a bénéficié du dispositif ATU/Post-ATU entre octobre 2013 et novembre 2014. Les autres molécules utilisées dans le traitement contre le VHC, notamment Daklinza® et Olysio® et Harvoni® sont entrées dans le dispositif courant 2014 et ont généré des dépenses jusqu'à mi-2015 ».
* 18 Opdivo® a bénéficié de l'ATU à partir de décembre 2014. Après une montée en charge en 2015 avec 47 M€ de remboursements versés, Opdivo® représente plus de la moitié des dépenses d'ATU pour 2016, soit 210 M€. Il est le premier contributeur à la croissance en 2016. En 2015, les remboursements de l'anti-PD1 Keytruda®, entrée dans le dispositif ATU en août 2014, s'élevaient à 37 M€. Il contribue également fortement à la croissance observée en 2016, puisqu'il est le 4e contributeur avec une hausse de ses dépenses de 24,5 M€ (comptes de la sécurité sociale, septembre 2017).
* 19 Il s'agissait d'une contribution transitoire, mise en place pour les années 2014 à 2016, à la charge des entreprises exploitant des médicaments dédiés au traitement de l'hépatite C. Le principe en était le suivant : en cas de dépassement d'un montant de chiffre d'affaires fixé par la loi de financement issu de l'exploitation des médicaments destinés au traitement l'hépatite C et d'un taux de croissance supérieur à 10 % du chiffre d'affaires de ces produits, le mécanisme de taxation se déclenchait. Le mécanisme W a été déclenché en 2014 comme en 2015, pour une contribution atteignant respectivement 282 et 11 millions d'euros.
* 20 Institut national du cancer, « Le prix des médicaments anticancéreux », mai 2017.
* 21 Observatoire du Cancer, Institut Curie - Viavoice, « Coûts des traitements innovants contre le cancer : perspectives d'un système en danger », 2017.
* 22 Loi n° 2011-2012 du 29 décembre 2011 relative au renforcement de la sécurité sanitaire du médicament et des dispositifs médicaux.
* 23 Loi n° 2013-1203 du 23 décembre 2013 de financement de la sécurité sociale pour 2014.
* 24 Article L. 162-16-5-2 du code de la sécurité sociale.
* 25 Cf. partie II.
* 26 Le British Medical Journal (BMJ), en conclusion d'un article d'octobre 2017 intitulé « Availability of evidence of benefits on overall survival and quality of life of cancer drugs approved by European Medicines Agency : retrospective cohort study of drug approvals 2009-2013 » et signé par Courtney Davis, Huseyin Naci, Evrim Gurpinar, Elita Poplavska, Ashlyn Pinto,Ajay Aggarwal, souligne par ailleurs que la plupart des médicaments anticancéreux approuvés par l'EMA de façon précoce, sur des critères intermédiaires, et mis sur le marché entre 2009 et 2013 n'ont pas apporté la preuve qu'ils amélioraient la survie globale ou la qualité de vie. Cet article a été porté à la connaissance de vos rapporteurs par les services ministériels.
* 27 Sur ce point, voir partie II. Le rapport d'activité de la HAS pour 2016 indique que sur 750 spécialités pharmaceutiques ayant fait l'objet d'un avis rendu au cours de l'année, seuls 25 médicaments (soit 3,3 % d'entre eux) ont été reconnus comme apportant une avancée thérapeutique par rapport aux traitements existants. Sur ces 25 produits, 6 seulement ont obtenu un ASMR de niveau II ou III, les 19 autres ayant reçu une ASMR IV.
* 28 En application de l'article l. 5121-12 du code de la santé publique.
* 29 Le régime des PUT est défini à l'article R. 5121-70 du code de la santé publique.
* 30 Bernard Bégaud, Dominique Polton et Franck von Lennep, « Les données de vie réelle, un enjeu majeur pour la qualité des soins et la régulation du système de santé. L'exemple du médicament », Rapport réalisé à la demande de la ministre de la santé Marisol Touraine, mai 2017.
* 31 Loi n° 2011-2012 du 29 décembre 2011 relative au renforcement de la sécurité sanitaire du médicament et des produits de santé.
* 32 Décret n° 2012-742 du 9 mai 2012 relatif aux recommandations temporaires d'utilisation des spécialités pharmaceutiques modifié et décret n° 2012-740 du 9 mai 2012 relatif à la prise en charge dérogatoire par l'assurance maladie des spécialités pharmaceutiques bénéficiant d'une recommandation temporaire d'utilisation ou de certains produits et prestations.
* 33 Loi n° 2016-1827 du 23 décembre 2016 de financement de la sécurité sociale pour 2017.
* 34 Code communautaire relatif aux médicaments à usage humain, transposé à l'article L. 5121-8 du code de la santé publique.
* 35 D'après une étude internationale de l'INCa publiée en janvier 2018 sur l'accès aux innovations médicamenteuses en cancérologie, les États-Unis se positionnent en effet souvent en premier dans les décisions d'autorisation de mise sur le marché, devant l'agence européenne.
* 36 Ces membres titulaires ayant voix délibérative sont nommés par décision du collège de la HAS pour une durée de trois ans renouvelable deux fois ; par ailleurs, sept membres suppléants assistent aux séances avec voix consultative ; des membres ont enfin une voix consultative (le directeur de la sécurité sociale, le directeur général de la santé, le directeur général de l'offre de soins, le directeur général de l'ANSM, les directeurs de la Cnam et de la MSA, ou leurs représentants).
* 37 Cette commission est composée de 33 membres titulaires (professionnels de santé, économistes, épidémiologistes, sociologues et autres disciplines des sciences humaines et sociales, membre d'une association de patients et d'usagers) ayant voix délibérative.
* 38 Rapport de mandature de la Commission évaluation économique et santé publique (2015-2017), HAS, février 2018.
* 39 Direction de la sécurité sociale, direction générale de la santé, direction générale de la concurrence, de la consommation et de la répression des fraudes, direction générale des entreprises et, avec voix consultative, direction générale de l'offre de soins.
* 40 « Le médicament : à quel prix ? », rapport d'information n° 739 (2015-2016), fait par MM. Gilbert Barbier et Yves Daudigny, au nom de la commission des affaires sociales, Sénat, 29 juin 2016.
* 41 Rapport sur l'application des lois de financement de la sécurité sociale (Ralfss), Chapitre VIII - « La fixation du prix des médicaments : des résultats significatifs, des enjeux toujours majeurs d'efficience et de soutenabilité, un cadre d'action à fortement rééquilibrer », Cour des comptes, septembre 2017.
* 42 Cf. partie III ci-après.
* 43 Directive 89/105/CEE ou Directive Transparence, qui harmonise les délais réglementaires d'accès au marché en Europe. Ce délai s'applique entre la demande d'inscription sur les listes des spécialités prises en charge et les décisions de prise en charge ou de prix.
* 44 Ce délai est apprécié du dépôt du dossier par les industriels (parallèlement au dépôt de ce dossier à la commission de transparence) à sa conclusion (par la clôture du dossier ou la parution du prix au Journal Officiel), incluant donc une phase d'évaluation par la HAS dont l'évaluation est toutefois déconnectée des données de la HAS et bien inférieure (soit 80 jours pour les médicaments non génériques et 29 jours en moyenne).
* 45 Sur un échantillon de 935 demandes de première inscription auxquelles le CEPS a répondu en 2016, dont 65 % relatives à des médicaments génériques.
* 46 Sur 64 dossiers de médicaments à l'hôpital traités par le CEPS en 2016, dont 39 concernaient des médicaments rétrocédables et 25 des médicaments facturés en sus des prestations d'hospitalisation.
* 47 European Federation of Pharmaceutical Industries Associations (EFPIA).
* 48 L'EFPIA a mis en place le « Patients W.A.I.T. indicator », montrant le taux de disponibilité des nouveaux médicaments pour les patients à travers l'Europe ainsi que le délai moyen ou médian entre l'AMM et l'accès au patient.
* 49 Committee for Medicinal Products for Human Use (CHMP).
* 50 Rapport sur les charges et produits de l'Assurance maladie pour 2018, Cnamts, juillet 2017.
* 51 On peut toutefois noter que la dépense de médicaments remboursés connaît en Allemagne sur la période 2012-2016 une croissance de + 4,7 % en moyenne par an (d'après la Cour des comptes, Ralfss 2017). D'après le rapport d'activité 2016 du CEPS, le marché des médicaments remboursables progresse en France de + 2,6 % en 2016 contre + 0,6 % en 2015, la croissance étant principalement tirée par les produits en ATU.
* 52 CEPS, rapport d'activité 2016, annexe 4 (les méthodes de fixation du prix du médicament).
* 53 La précédente lettre d'orientation ministérielle de 2013 prévoyait que le prix d'un médicament avec une ASMR IV ne devait « pas entraîner de surcoût pour l'assurance maladie » ; depuis 2016, l'absence de surcoût n'est plus considérée sur la base d'une moyenne pondérée des comparateurs mais par rapport au coût net du comparateur le moins cher.
* 54 CEPS, rapport d'activité pour l'année 2016.
* 55 La lettre d'orientation ministérielle du 17 août 2016 au président du CEPS définit comme suit le recours à ces comparateurs : « Vous prendrez en considération l'ensemble des comparateurs cités par la HAS mais en l'absence de tels comparateurs, ou si cela peut se justifier de manière appropriée, vous utiliserez des comparateurs économiquement pertinents au regard des connaissances médicales avérées qu'il s'agisse de produits de santé, d'actes ou de prestations. »
* 56 « Innovation médicamenteuse en cancérologie - étude internationale sur la définition et l'accès à l'innovation », Institut national du cancer, janvier 2018.
* 57 Le CEPS a mis en place début 2015 un « Comité de prospective des innovations médicamenteuses » qui ne paraît pas toutefois répondre actuellement à ce besoin.
* 58 « Innovation en santé : soignons nos talents », Institut Montaigne, mars 2018.
* 59 A noter toutefois qu'une entreprise a la possibilité d'exposer ses arguments lors d'une phase contradictoire en cas de désaccord avec l'appréciation de la commission de la transparence.
* 60 Rapport sur la réforme des modalités d'évaluation des médicaments, remis à la ministre en charge de la santé par Mme Dominique Polton, novembre 2015.
* 61 Depuis l'accord-cadre de 2015 entre le Leem et le CEPS, des réserves méthodologiques émises par la commission compétente de la HAS sont susceptibles de conduire à l'inéligibilité de certains médicaments à la garantie de prix européen.
* 62 D'après une étude internationale de l'INCa précitée, 27 décisions de ce type ont été rendues par la G-BA depuis 2011 (données 2015).
* 63 D'après le rapport de l'Institut Montaigne sur l'innovation en santé, mars 2018.
* 64 Voir récemment l'avis du Conseil économique, social et environnemental (CESE) « Prix et accès aux traitements médicamenteux innovants », n° 2017-04, présenté par Mme Catherine Pajarès y Sanchez et M. Christian Saout, 25 janvier 2017.
* 65 Dans son rapport « charges et produits » pour 2018, l'Assurance maladie proposait ainsi d' « adapter les modalités de fixation des prix à la dynamique des médicaments innovants ».
* 66 Le principe de contrats de performance est prévu à l'article 12 de l'accord-cadre de 2015 entre le CEPS et le Leem : « Au cas où les modalités de fixation des prix de droit commun ne permettraient pas de trouver un accord, le prix de certains médicaments peut être fixé, à la demande du Comité ou de l'entreprise, conditionnellement au résultat de la performance constatée en vie réelle. »
* 67 « Les données de vie réelle, un enjeu majeur pour la qualité des soins et la régulation du système de santé - L'exemple du médicament », Pr Bernard Bégaud, Dominique Polton et Franck von Lennep, mai 2017.
* 68 Modèle PRM, pour Personal reimbursement model.
* 69 En l'absence de RTU dans l'indication ou les conditions d'utilisation considérées, l'article L. 5121-12-1 du code de la santé publique prévoit que la prescription hors AMM n'est possible « qu'en l'absence d'alternative médicamenteuse appropriée disposant d'une autorisation de mise sur le marché ou d'une autorisation temporaire d'utilisation et sous réserve que le prescripteur juge indispensable, au regard des données acquises de la science, le recours à cette spécialité pour améliorer ou stabiliser l'état clinique de son patient. » Les conditions d'information du patient sont par ailleurs strictement encadrées.
* 70 Cf. partie I.
* 71 Ces référentiels viendraient rétablir en quelque sorte ceux supprimés par l'article 81 de la loi de financement de la sécurité sociale pour 2016, qui relevaient de l'INCa pour l'oncologie et de l'ANSM dans les autres champs thérapeutique.
* 72 Source : rapport d'activité 2016 du CEPS, décembre 2017.
* 73 Permettant notamment de traiter la polyarthrite rhumatoïde et des maladies inflammatoires chroniques de l'intestin.
* 74 Loi n° 2013-1203 du 23 décembre 2013 de financement de la sécurité sociale pour 2014 (article 51).
* 75 Recommandation n° 2010-25 du 18 novembre 2010 du conseil de l'hospitalisation.
A l'issue d'un contentieux intenté par deux laboratoires, le Conseil d'État a jugé que ces critères devaient être fixés par un texte règlementaire et non plus seulement dans une recommandation, pour répondre aux exigences de la directive européenne dite directive « transparence ».
* 76 Cette radiation a été effective le 1 er mars 2018.
* 77 Communiqué de presse de l'association Renaloo, « Les patients greffés du rein toujours privés du traitement antirejet le plus efficace ! », 4 février 2016.
* 78 « Améliorer la pertinence des soins : un enjeu majeur pour notre système de santé », rapport d'information n° 668 (2016-2017) de M. Jean-Marie Vanlerenberghe, rapporteur général, au nom de la mission d'évaluation et de contrôle de la sécurité sociale et de la commission des affaires sociales, Sénat, 20 juillet 2017.
* 79 Loi n° 2017-1836 du 30 décembre 2017 de financement de la sécurité sociale pour 2018.
* 80 Article L. 162-31-1 du code de la sécurité sociale, dont les modalités ont été précisées par le décret n° 2018-125 du 21 février 2018.
* 81 Environ 100 000 patients reçoivent une chimiothérapie à base de 5-FU en France chaque année, selon l'Institut national du cancer (INCa).
* 82 C'est le cas des patients présentant une carence en dihydropyrimidine deshydrogénase (DPD), enzyme permettant le métabolisme et la dégradation dans l'organisme des fluoropyrimidines. De 3 à 10 % des patients présentent un déficit partiel en DPD et entre 0,1 et 0,5% un déficit total.
* 83 Instruction n° DGOS/PF4/2015/258 du 31 juillet 2015. Avant 2015, la liste des actes pris en charge par une dotation annuelle spécifique (dotation constante chaque année de 250 millions d'euros hors actes de génétique pris en charge par la dotation MERRI de 130 millions d'euros) était mise à jour par le CHU de Montpellier et appelée « grille de Montpellier ».
* 84 Missions d'intérêt général d'enseignement, de recherche, de rôle de référence et d'innovation.
* 85 Le coût d'un test est d'environ 1 000 à 2 000 euros ; ce test est à faire plusieurs fois dans la vie d'une tumeur.
* 86 Loi n° 88-1138 du 20 décembre 1988 relative à la protection des personnes qui se prêtent à des recherches biomédicales.
* 87 « Principes éthiques applicables aux recherches médicales sur des sujets humains », Déclaration d'Helsinki de l'Association médicale mondiale (AMM), 1964.
* 88 Loi n° 2004-806 du 9 août 2004 relative à la politique de santé publique.
* 89 Directive 2001/20/CE du Parlement européen et du Conseil du 4 avril 2001 concernant le rapprochement des dispositions législatives, réglementaires et administratives des États membres relatives à l'application de bonnes pratiques cliniques dans la conduite d'essais cliniques de médicaments à usage humain.
* 90 Loi n° 2012-300 du 5 mars 2012 relative aux recherches impliquant la personne humaine.
* 91 Loi n° 2016-41 du 26 janvier 2016 de modernisation de notre système de santé.
* 92 Ordonnance n° 2016-800 du 16 juin 2016 relative aux recherches impliquant la personne humaine.
* 93 Décret n° 2016-1537 du 16 novembre 2016 relatif aux recherches impliquant la personne humaine.
* 94 Article L. 1123-7 du code de la santé publique.
* 95 « Essais précoces en cancérologie, éthique et justice. La lettre du cancérologue, 2012, XXI (10), pp. 514-518.
* 96 https://www.gustaveroussy.fr/fr/essais-precoces (consulté le 13 juin 2018).
* 97 Leem, 8 ème enquête « Attractivité de la France pour la recherche clinique internationale », février 2017. Cette étude porte sur les essais interventionnels à promotion industrielle enregistrée sur le site www.clinicaltrials.gov pour les années 2014 et 2015.
* 98 Document de travail des services de la commission ; résumé du rapport d'analyse d'impact relatif à la révision de la directive 2001/20/CE sur les essais cliniques accompagnant le document Proposition de règlement du Parlement européen et du Conseil relatif aux essais cliniques de médicaments à usage humain et abrogeant la directive 2001/20/CE.
* 99 Selon les réponses apportées tardivement le 12 juin 2018 au questionnaire de vos rapporteurs, pour 2017, la répartition entre les différents domaines thérapeutiques est la suivante : 54% pour l'oncologie, hématologie, transplantation, néphrologie, thérapie cellulaire, produits sanguins et radio-pharmaceutiques, 19% pour la cardiologie, rhumatologie, stomatologie, endocrinologie, gynécologie, urologie, pneumologie, ORL, allergologie, 11% pour la neurologie, psychiatrie, anesthésie, antalgie, ophtalmologie, stupéfiants, psychotropes et médicaments des addictions, 16% pour les vaccins, médicaments anti-infectieux, en hépato-gastro-entérologie, dermatologie, de thérapie génique et maladies métaboliques rares.
* 100 Leem, 8ème enquête « Attractivité de la France pour la recherche clinique internationale », février 2017. Cette étude porte sur les essais interventionnels à promotion industrielle enregistrée sur le site www.clinicaltrials.gov pour les années 2014 et 2015.
* 101 Réponses au questionnaire de vos rapporteurs.
* 102 Règlement UE n° 536/2014 du Parlement européen et du Conseil du 16 avril 2014 abrogeant la directive 2001/20/CE du Parlement et du Conseil du 4 avril 2001 concernant le rapprochement des dispositions législatives, réglementaires des États membres relatives à l'application de bonnes pratiques cliniques dans la conduites d'essais cliniques de médicaments à usage humain.
* 103 Ordonnance n° 2016-800 du 16 juin 2016 relative aux recherches impliquant la personne humaine.
* 104 Proposition de loi relative à l'expertise des comités de protection des personnes, présentée par MM. Cyrille Isaac-Sibille, Philippe Berta et les membres du groupe Modem et apparentés, députés, adoptée en première lecture par l'Assemblée nationale le 9 mai 2018.
* 105 Depuis le 18 décembre 2017, cette cellule est chargée d'instruire les essais cliniques précoces portant sur les médicaments uniquement : il s'agit des essais de première administration chez l'homme et des essais visant à améliorer la connaissance initiale chez l'homme des profils de tolérance, sécurité, pharmacocinétique et pharmacodynamique. Ce sont des essais de phase I ou de phase I et II.
* 106 L'ANSM indique que : « Un conseiller médical a rejoint la DPAI en charge de la cellule. Son rôle est d'accompagner l'évaluation des essais à différentes étapes du traitement, notamment sur le plan de la mise en perspective des questions posées au cours de l'évaluation au regard de l'enjeu thérapeutique que constitue l'accès à un nouveau traitement dans des pathologies ou le pronostic vital est engagé. Des ressources d'autres directions sont mobilisées en tant que de besoin pour un traitement adapté et prioritaire de ce type d'essais cliniques. »
* 107 Réponses au questionnaire de vos rapporteurs.
* 108 Décret n° 2016-1538 du 16 novembre 2016 relatif à la convention unique pour la mise en oeuvre des recherches à finalité commerciale impliquant la personne humaine dans les établissements de santé, les maisons et les centres de santé.
* 109 Amendement n° 424 rect. présenté par M. Savary et plusieurs de ses collègues du groupe Les Républicains, 13 novembre 2017.
* 110 Réponses au questionnaire de vos rapporteurs.
* 111 Le « Right to try Act » a été adopté par le Congrès américain en mai 2018.