







Rapport n° 791 (2010-2011) de M. Claude BIRRAUX, député, fait au nom de l'Office parlementaire d'évaluation des choix scientifiques et technologiques, déposé le 14 septembre 2011
Disponible au format PDF (4,4 Moctets)
N° 3723 N° 791
____ ___
ASSEMBLÉE NATIONALE SÉNAT
CONSTITUTION DU 4 OCTOBRE 1958
TREIZIÈME LÉGISLATURE SESSION EXTRAORDINAIRE DE 2010 - 2011
____________________________________ ___________________________
Enregistré à la présidence de l'Assemblée nationale Enregistré à la présidence du Sénat
le 15 septembre 2011 le 14 septembre 2011
________________________
OFFICE PARLEMENTAIRE D'ÉVALUATION
DES CHOIX SCIENTIFIQUES ET TECHNOLOGIQUES
________________________
RAPPORT
sur
LES SAUTS TECHNOLOGIQUES EN MÉDECINE
Compte rendu de l'audition
publique du 27 janvier 2011
et de la présentation des conclusions, le
28 juin 2011
Par M. Claude Birraux, Député.
__________ __________
Déposé sur le Bureau de l'Assemblée nationale Déposé sur le Bureau du Sénat
par M. Claude BIRRAUX, par M. Bruno SIDO,
Président de l'Office Premier Vice-Président
_________________________________________________________________________
SOMMAIRE
___
Discours d'ouverture de M. Claude Birraux, député, président de l'OPECST 5
Propos introductif de M. Laurent Degos, ancien président de la Haute autorité de santé 7
Première
table ronde : Les thérapies cellulaires -
Présidence de
Mme Emmanuelle Prada-Bordenave, directrice générale de
l'Agence de biomédecine
11
Mme Emmanuelle Prada-Bordenave, directrice générale de l'Agence de la biomédecine 11
Mme Marina Cavazzana-Calvo, directrice du département de biothérapie à l'hôpital Necker 12
M. Philippe
Menasché, directeur de l'unité « Thérapie
cellulaire en pathologie cardio-vasculaire »
à
l'hôpital européen Georges Pompidou 14
M. Luc Sensebé, directeur médical et scientifique de l'Établissement français du sang (EFS) de la région Centre 18
M. Luc Douay, directeur médical et scientifique de l'Établissement français du sang (EFS) d'Île-de-France 20
Deuxième
table ronde : les actes médicaux -
Présidence de
M.
Jean Marimbert, directeur général de l'agence française de
sécurité sanitaire des produits de santé
(AFSSAPS)
33
M. Jean Marimbert, directeur général de l'AFSSAPS 33
M. Luc Mallet, psychiatre, Centre de recherche de l'Institut du cerveau et de la moëlle épinière 34
Mme Jacqueline
Mandelbaum, biologiste de la reproduction, chef du service d'histologie,
biologie de la reproduction / CECOS (Centre d'étude et de conservation
des oeufs et du sperme humains) de l'hôpital
Tenon, membre du Conseil
d'orientation de l'Agence de biomédecine 36
M. Elias
Zerhouni, président Monde, recherche et développement, en charge
des médicaments et
des vaccins, Sanofi-Aventis 42
Troisième
table ronde : les dispositifs médicaux -
Présidence de
M. Jean-Michel Dubernard, membre du collège de la haute
autorité
de santé
53
M. Jean-Michel Dubernard, membre du Collège de la Haute autorité de santé 53
M. Alain
Carpentier, cardiologue, hôpital européen Georges-Pompidou,
président de
l'Académie des sciences 54
M. Ugo Amaldi, physicien à l'Organisation européenne pour la recherche nucléaire (CERN). 57
M. José-Alain Sahel, directeur de l'Institut de la vision 62
Discours de clôture de M. Claude Birraux, député, président de l'OPECST 75
AUDITION PUBLIQUE DU 27 JANVIER 2011
DISCOURS D'OUVERTURE DE M. CLAUDE BIRRAUX, DÉPUTÉ, PRÉSIDENT DE L'OPECST
M. le président Claude Birraux . L'idée de cette audition publique, dont je me félicite, m'a été suggérée par le professeur Jean-Michel Dubernard. Son thème montre, si besoin était, la place éminente qu'occupent les questions médicales et celles touchant à l'innovation technologique dans les travaux de l'Office.
Depuis le début de cette législature, l'Office s'est penché sur la mutation des virus et la gestion des pandémies - notamment celle de la grippe H1N1 -, le dossier médical personnel, les apports de la science et de la technologie à la compensation du handicap et la radiothérapie. Le législateur, reconnaissant son expertise, l'a chargé d'évaluer l'application de la loi de bioéthique du 6 août 2004, à la révision de laquelle l'Assemblée nationale procède actuellement. Nos collègues Alain Claeys et Jean-Sébastien Vialatte ont ainsi rendu deux rapports consacrés l'un à cette évaluation des lois de bioéthique, l'autre à la recherche sur les cellules souches. Tous deux viennent en outre d'être chargés par l'Office d'une étude sur l'impact des nouvelles technologies dans les neurosciences et la neuro-ingénierie.
Pour ce qui est de l'innovation technologique, je suis fier de pouvoir dire que l'Office en a, à plusieurs reprises et de manière approfondie, abordé les différents aspects. Mon collègue Jean-Yves Le Déaut et moi-même venons de nous voir confier une étude sur le thème de l'innovation à l'épreuve des peurs et des risques majeurs.
Cette future étude comme cette audition publique interviennent à un moment où une grave crise sanitaire affectant le secteur du médicament risque d'accroître les peurs de l'opinion publique et sa méfiance envers les traitements et la médecine - ne parlons même pas de tous les délires auxquels donne lieu internet. Au-delà de ce hasard de l'actualité, se retrouve la même ambition d'analyser les conditions nécessaires pour permettre à l'innovation de jouer un rôle majeur, en tirant les leçons aussi bien des échecs que des succès.
Les processus d'innovation sont hétérogènes. Il existe toutefois une propension en médecine, comme dans d'autres domaines, à gommer cette hétérogénéité. La Cour de cassation fait ainsi observer, dans un commentaire au sujet des lois de bioéthique, que les innovations technologiques ont touché tous les secteurs de la médecine. Ce n'est pas faux. Pour autant, certains sauts technologiques sont mort-nés, d'autres n'ont pu avoir lieu que très difficilement, d'autres enfin n'ont tout simplement pas pu se réaliser dans notre pays et se sont développés à l'étranger.
Il en va ainsi de l'hadronthérapie, destinée à traiter les cancers inopérables et radiorésistants. Si j'ai tenu à ce que son inventeur, le professeur Ugo Amaldi, spécialiste de physique des particules, docteur honoris causa de la faculté de médecine de Lyon, participe aujourd'hui à nos travaux, c'est pour qu'il nous dise les obstacles qu'il a rencontrés en France pour y développer cette innovation, issue de travaux du CERN (Centre européen de recherche nucléaire), et qui l'ont conduit à retourner dans son pays natal. Il était venu me voir en août 2005 quand les ministères français concernés lui répondaient qu'un grand projet, ÉTOILE, était en cours en Rhône-Alpes et qu'il était hors de question d'en soutenir un second. Le professeur Amaldi est alors parti ouvrir un premier centre d'hadronthérapie à Pavie. Un second est depuis ouvert à Trente et d'autres projets sont en cours, notamment à Rome. Pendant ce temps en France, ÉTOILE patine et le centre de traitement ne devrait pas pouvoir ouvrir avant 2016: il a fallu relancer un appel d'offres, le premier ayant été infructueux après qu'on a tenté un partenariat public-privé. La difficulté d'innover sur le plan médical en France tient au caractère administré de notre médecine. Le premier « TEP scan » 1 en France a été installé dans une clinique équine en Normandie : c'est tout dire !
Le parcours du professeur Elias Zerhouni, qui traitera tout à l'heure des grandes tendances de l'innovation biomédicale au 21 ème siècle, montre fort heureusement que la fuite des cerveaux n'est pas une fatalité. Après avoir effectué une très brillante carrière aux États-Unis, le professeur Zerhouni est revenu en France où il a donné, il y a quelques jours, sa leçon inaugurale au Collège de France sur ce même thème qu'il nous exposera.
Quoi qu'il en soit, il importe de mener une politique de l'innovation technologique efficace pour prévenir la fuite des cerveaux et favoriser le développement des innovations en France plutôt qu'à l'étranger. Les industries des instruments médicaux sont de haute technologie. D'après un rapport de mai 2009 de Coe-Rexecode sur la compétitivité de l'industrie mécanique française, la valeur ajoutée de ces industries s'élevait en 2007 à près de 10 milliards de dollars sur un total de 70,2 milliards de dollars pour l'ensemble de l'industrie mécanique. La France se classe au huitième rang mondial pour les exportations mondiales d'instruments médicaux, avec 4,5% de part de marché, et au troisième rang européen, après l'Allemagne et la Suisse.
Le Gouvernement a pris la mesure de l'exigence de compétitivité en créant Medicen, pôle de compétitivité mondial dans le domaine des hautes technologies pour la santé et les nouvelles thérapies. Cette décision va dans le bon sens. D'après un article de The Economist paru la semaine dernière, qui reprend les conclusions d'une étude de Price WaterHouse Coopers sur la compétitivité en innovation technologique médicale, la France se situe à un niveau très honorable. Elle se classait en 2010 au cinquième rang mondial, juste après le Japon, et au troisième rang en Europe, après l'Allemagne et le Royaume-Uni, pour ce qui est des performances de compétitivité, calculées sur la base d'une série de 86 mesures concernant l'économie, la réglementation, l'organisation et le marché.
Sans sous-estimer ces bonnes nouvelles, ne nous cachons pas qu'il reste beaucoup à faire. D'après un rapport de Coe-Rexecode encore, la part de l'Allemagne dans les exportations mondiales d'instruments médicaux était, en 2007, trois fois supérieure à celle de la France, 13,6% contre 4,5%. Ce même rapport, ainsi que l'étude de Price WaterHouse Coopers, soulignent par ailleurs le dynamisme croissant de l'Asie.
L'ardente obligation qui incombe aux pouvoirs publics et aux industriels ne se limite pas à renforcer notre compétitivité économique. Il leur faut aussi lutter contre un climat d'obscurantisme et de méfiance envers la science, en montrant que l'innovation en matière médicale ne peut qu'être source de progrès et bénéfique à l'humanité.
Je forme le souhait que cette audition publique, ouverte à la presse, contribue à diffuser ce message.
PROPOS INTRODUCTIF DE M. LAURENT DEGOS, ANCIEN PRÉSIDENT DE LA HAUTE AUTORITÉ DE SANTÉ .
Je suis très heureux d'être parmi vous pour débattre d'un sujet tout à fait d'actualité. Tout progrès, dans sa phase initiale, comporte un bénéfice escompté et un risque imprévisible. Si le rapport bénéfice/risque d'un produit de santé pour un individu constitue le critère d'autorisation de mise sur le marché ou de retrait de celle-ci, son évaluation est beaucoup plus délicate lorsque le bénéfice est collectif et potentiel alors que le risque individuel, imprévisible, peut être immédiat et non négligeable. C'est là toute la problématique du saut technologique, moment où la société ignore encore le résultat d'un produit ou d'une pratique, mais porte beaucoup d'espoir en ses bienfaits tout en étant prête à en assumer les risques.
On ne pratiquerait pas de greffes aujourd'hui sans la persévérance des acteurs médicaux et l'insistance des patients, chaque greffé ayant au début espéré être le premier survivant. Il a fallu pas moins de 263 greffes du foie avant d'obtenir qu'un patient greffé survive plus d'une semaine ! Aujourd'hui, une agence sanitaire n'imposerait-elle pas l'arrêt de la pratique bien avant un tel nombre d'essais infructueux ? Il a de même fallu surmonter les effets indésirables provoqués par la chirurgie sous coelioscopie à ses débuts, alors qu'elle est aujourd'hui devenue la pratique courante, préférée à des techniques plus invasives. L'histoire des découvertes médicales est constituée d'actes souvent hors la loi, d'interdits contournés par les médecins, avec l'appui de leurs patients. Ainsi les premiers médecins qui prélevaient des greffons sur des patients en état de mort encéphalique mais dont le coeur battait encore pouvaient être poursuivis pour homicide volontaire. Cette situation a perduré de 1963, date du premier prélèvement de ce type, à 1968, où l'état de mort cérébrale fut officiellement reconnu par circulaire ministérielle. Aucun de ces praticiens n'a, fort heureusement, été poursuivi, mais c'est dire d'où on vient !
Peut-on proposer un traitement totalement novateur, présentant des risques inconnus pour l'individu sur lequel il est expérimenté, au profit d'un espoir de thérapie future, utile à la collectivité ? Il ne s'agit pas de conflit entre progrès et précaution ni sécurité, mais bien de relation entre progrès et éthique. Sur le plan individuel, le patient réclame que tout soit essayé pour le traiter, étant prêt à assumer un risque pour cette recherche d'efficacité, alors que sur le plan collectif, la sécurité passe avant l'efficacité. Du point de vue individuel, on ne peut volontairement faire courir des risques à une personne, tandis que du point de vue collectif, aucun progrès n'est possible sans essai dont on ne maîtrise pas les risques.
Dès 1984, le Comité consultatif national d'éthique (CCNE) relevait, dans son avis du 9 octobre, que le médecin « se trouve ainsi confronté, sur le plan de l'éthique, à deux impératifs. Le souci de l'intérêt de son patient lui enjoint de lui administrer le traitement considéré comme le meilleur dans l'état actuel des connaissances ; le souci du bien collectif, de la santé publique, lui dicte de faire en sorte que le traitement de son patient puisse contribuer au progrès de la thérapeutique. » Dans son avis du 26 septembre 2002, le CCNE soulignait que « le progrès médical s'est souvent fondé sur des rapports bénéfices/risques initialement asymétriques au détriment des bénéfices. La gestion de cette contradiction n'est possible que si la nécessité absolue et permanente de disposer de nouvelles molécules, idée partagée par les malades, leur famille et la société, s'accompagne de la conscience de cette asymétrie. Chaque malade doit pouvoir comprendre que toute thérapeutique innovante s'est toujours fondée sur de tels essais chez d'autres malades et n'est jamais issue de la seule expérimentation animale. En cette circonstance, le droit de la personne ne peut pas être mis en opposition avec le devoir de solidarité. La société dans son ensemble doit être consciente que l'exigence de la recherche peut conduire à privilégier parfois les intérêts de la communauté. Cependant, cette conscience même n'abolit jamais l'impératif majeur de respecter totalement cette personne qui, par sa maladie même, peut en effet venir en aide à l'humanité. »
Pour l'évaluation du rapport bénéfice collectif potentiel/risque individuel immédiat, la décision se fonde davantage sur des arguments sociétaux et d'éthique que de sécurité sanitaire. C'est pourquoi aux États-Unis par exemple, les autorisations pour des sauts technologiques comme les xénotransplantations, sont données par des « comités sociétaux » adossés aux universités, et non par des agences de sécurité sanitaire. Pour se prononcer, ces comités se fondent sur l'évaluation du futur bénéfice potentiel collectif par rapport au risque individuel immédiat et vérifient que la personne qui accepte ainsi de « venir en aide à l'humanité » a été parfaitement informée et a bien compris ce qu'on lui proposait. Si les agences de sécurité sanitaire défendent l'intérêt collectif en matière de santé et s'attachent, par fonction, à la sécurité sanitaire, le patient, lui, demande aux médecins de tout tenter pour lui, même s'il doit subir des effets secondaires majeurs. Ainsi la greffe de moelle, en dépit d'un risque de mortalité de 10 à 20%, est-elle très largement pratiquée.
Dans notre pays, l'Agence de la biomédecine comporte un conseil d'orientation qui joue ce rôle sociétal de conscience et de discernement collectifs pour la greffe, la procréation artificielle et la génétique. Ce pourrait être un modèle pour le futur. Ses avis sont cependant revus ensuite par l'Agence française de sécurité sanitaire des produits de santé, l'AFSSAPS, qui autorise ou non les essais suivant des règles strictes. On ne peut toutefois avoir deux visions de la sécurité, l'une pour les technologies bien connues, l'autre pour les nouveautés, au risque d'empêcher parfois certains sauts technologiques.
L'audition publique d'aujourd'hui doit nous éclairer. Comment définir le saut technologique ? Qui peut juger de son bien-fondé ? Qui l'autorise ? Qui apporte le discernement sociétal ? Qui évalue et suit l'évolution des actes et produits particulièrement innovants ? Comment ne pas entraver le progrès tout en respectant l'individu qui « vient en aide à l'humanité » en acceptant de prendre des risques importants parce qu'il en en escompte un bénéfice pour lui-même ? Autant de questions dont nous allons aujourd'hui utilement débattre, car certains peuvent ressentir un blocage dans les avancées possibles.
M. le président Claude Birraux . Je vous remercie de ce propos liminaire. Nous croyons tous au progrès scientifique et technologique, il me paraît important de le rappeler car nos propos sont souvent déformés. J'ai le souvenir d'un entretien il y a quelques années avec un journaliste dont je me suis aperçu que le seul objectif était de me faire dire qu'il fallait décréter un moratoire sur toutes les recherches, car celles-ci ne conduisaient qu'à des catastrophes. Durant plus de trois heures, il a désespérément essayé de me le faire dire. J'ai résisté, lui suggérant même d'aller proposer un tel moratoire à des malades du sida ! De toute façon, le corps social se rebellerait car il n'accepterait pas votre diktat, ai-je conclu.
Nous allons maintenant ouvrir notre première table ronde.
PREMIÈRE TABLE
RONDE : LES THÉRAPIES CELLULAIRES
Présidence de
Mme Emmanuelle Prada-Bordenave,
directrice générale de
l'Agence de biomédecine
Mme Emmanuelle Prada-Bordenave, directrice générale de l'Agence de la biomédecine . L'Agence de la biomédecine est, entre autres, chargée de la régulation des greffes d'organes, de tissus et de cellules. C'est un domaine où depuis cinquante ans dans notre pays, ont été accomplis des sauts technologiques majeurs, accompagnés et salués par les patients et l'ensemble de la société française. L'histoire de la greffe en France, c'est celle d'une adhésion forte entre le peuple français et les grands noms de la médecine qui les ont réalisées.
L'Agence accompagne et encadre donc les activités de thérapie cellulaire, essentiellement les greffes de moelle osseuse. Elle a aussi pour rôle de contrôler les recherches sur l'embryon et les cellules souches embryonnaires. Elle porte une attention constante aux recherches en cours, afin qu'en résultent des progrès thérapeutiques majeurs emportant l'adhésion de nos concitoyens, qui sont solidaires des patients et qui, tous, au fond d'eux-mêmes admirent les chercheurs qui rendent possibles ces avancées.
Nous allons successivement entendre, au cours de cette table ronde, Mme Marina Cavazzana-Calvo, figure mondiale de la thérapie génique, qui a conduit plusieurs essais cliniques dans le traitement du déficit immunitaire combiné sévère ; le Pr Philippe Menasché, qui fut l'un des pionniers de la thérapie cellulaire et va bientôt déposer une demande d'autorisation pour utiliser dans le traitement de l'insuffisance cardiaque, des cardiomyocytes différenciés à partir de cellules souches embryonnaires humaines ; le Pr Luc Sensebé, spécialiste des cellules souches mésenchymateuses, qui étudie notamment les aspects fondamentaux, cliniques et translationnels, des cellules souches adipocytaires et de la moelle dans la perspective de thérapies cellulaires ; enfin, le Pr Luc Douay qui a conduit de nombreux travaux sur la différenciation des cellules souches en cellules sanguines, en particulier en érythrocytes, et a été l'auteur en 2005 d'une publication majeure sur la production de ces globules rouges.
Les médias relaient tous ces progrès, présentant parfois comme acquis ce qui en est encore au stade de l'essai. Il est donc particulièrement intéressant d'entendre ces chercheurs de renommée mondiale nous dire quels obstacles ils ont rencontrés, mais aussi leur volonté résolue, en dépit des difficultés, d'aboutir à des avancées au bénéfice des patients.
Mme Marina Cavazzana-Calvo, directrice du département de biothérapie à l'hôpital Necker . Je remercie les organisateurs de cette journée de leur invitation. Je suis à la fois un exemple et un contre-exemple de la fuite des cerveaux évoquée par le président Birraux dans son propos liminaire. Italienne, j'ai pu mener en France des travaux que je n'aurais vraisemblablement pas pu conduire dans mon université d'origine de Padoue. Mais il me faut aujourd'hui retourner en Italie pour étendre les essais cliniques car je n'ai plus les moyens nécessaires pour ce développement en France.
Quatre essais cliniques de thérapie génique sont en cours à l'hôpital Necker, ce qui en fait le premier centre de thérapie génique au monde. Cette thérapie représente un défi complexe. Il s'agit d'insérer les bons gènes dans les bonnes cellules, de faire en sorte qu'ils ne s'y expriment ni trop faiblement ni trop fortement, d'en obtenir une expression prolongée et régulée, et, comme on l'exige de nous au nom du principe de précaution, d'éviter toute toxicité. Celle-ci peut résulter d'une mutagenèse insertionnelle, car le vecteur s'intègre d'une façon semi-alétoire dans l'ADN des cellules, d'une réaction inflammatoire due au virus vecteur ou d'une réponse immunologique contre la protéine fabriquée ex novo dans les cellules corrigées.
Pourquoi avons-nous choisi, à la fin des années 90, de nous attacher au traitement du déficit immunitaire combiné sévère lié au chromosome X ? D'une part, sur un plan éthique, il s'agissait bien d'une urgence thérapeutique, les enfants atteints de cette maladie étant voués à mourir très peu de temps après leur naissance en l'absence de traitement. Leur thymus ne produisant pas de globules blancs, en particulier de lymphocytes T, ils ne peuvent en effet se défendre contre aucune infection. D'autre part, après plus de dix ans de travaux conduits avec le professeur Alain Fischer et le professeur Salima Hcein-Bet-Abina, nous avions acquis la certitude que s'il y avait bien une maladie au traitement de laquelle pouvait s'appliquer la thérapie génique, c'était celle-là.
Le principe de cette thérapie consiste à modifier un virus - car seuls les virus sont capables d'insérer une information génétique nouvelle au coeur du génome - d'une part pour lui ôter sa capacité de reproduction, d'autre part y introduire, par des méthodes de biologie moléculaire, les gènes thérapeutiques qui prendront place ultérieurement dans le génome des cellules à traiter et permettront d'y fabriquer la protéine qui fait défaut.
Avant d'en arriver aux essais cliniques, le parcours a été assez long si on se place de notre point de vue ou de celui des malades, mais relativement court par rapport au développement d'un produit pharmaceutique. Nous avons d'abord démontré in vitro qu'il était possible de faire fabriquer ainsi la protéine manquante, de manière stable ; ensuite nous avons démontré, en introduisant le gène manquant dans les cellules souches de l'individu malade, la possibilité de restaurer leur capacité de différenciation en globules blancs. Des souris reproduisant la maladie humaine ont été générées par thérapie génique, démontrant ainsi que l'application thérapeutique était possible et ne présentait pas de toxicité.
Il nous a fallu ensuite passer du protocole expérimental au protocole clinique, non plus chez l'animal mais chez l'homme, ce qui impose de travailler avec des matériels approuvés par l'AFSSAPS, effectuer des expériences à l'échelle clinique et codifier toutes les procédures pour être en conformité avec la législation française. Nous avons commencé nos premiers travaux dès la découverte du gène responsable de la maladie en 1994, mais n'avons pu lancer les premiers essais cliniques qu'en 1999.
Quel en est le principe ? On prélève sous anesthésie générale de la moelle osseuse chez un enfant malade. On en purifie les cellules souches qui sont placées pendant cinq jours au contact du virus qui contient le gène thérapeutique, puis réinjectées à l'enfant par voie intra-veineuse. Il faut ici souligner que la France est un exemple en Europe et dans le monde pour le soutien sociétal et financier qui y est accordé à l'activité de greffe. Sans l'expérience accumulée depuis une vingtaine d'années grâce aux greffes de moelle osseuse, expérience à la fois clinique et biologique, jamais notre essai n'aurait pu voir le jour.
Quels ont été nos résultats ? Rappelons que les enfants présentaient, en l'absence de traitement, un pronostic sombre caractérisé par un décès dans la première année de vie. Aujourd'hui, dix-huit des vingt enfants, traités à Paris et à Londres, mènent une existence tout à fait normale, sans autre traitement. Même en cas d'histocompatibilité, la greffe de moelle osseuse présente un risque de mortalité de 10 à 20%, soit exactement le même que celui observé à long terme avec cette option thérapeutique. Un seul décès lié directement à la thérapie génique a en effet été à déplorer.
Nous avons néanmoins dû suspendre notre essai en 2002. Alors que cela ne s'était pas produit chez la souris et que cela n'était aucunement prédictible à l'issue de tous les essais pré-cliniques, quatre des dix enfants traités ont développé une leucémie. L'essai a donc été suspendu en accord avec l'AFSSAPS, le temps d'analyser le rapport bénéfice/risque et de chercher comment réduire au maximum le risque d'oncogenèse.
De 2002, date d'arrêt de l'essai, à 2010, nous avons mené diverses recherches qui nous ont conduits à changer de vecteur mais aussi à étudier si cette thérapie ne pourrait pas être utilisée également pour d'autres maladies, dont la bêta-thalassémie et la drépanocytose, première maladie génétique dans le monde, voire l'adrénoleucodystrophie liée au chromosome X, maladie neurologique dévastatrice du jeune enfant.
Nous avons obtenu en 2010 une nouvelle autorisation d'essai clinique international multicentrique - il est mené également au Great Ormond Street Hospital de Londres et au Children's Hospital de Boston - et avons inclus les premiers patients dès cette année.
Mme Emmanuelle Prada-Bordenave . Nous aurons l'occasion de revenir sur les obstacles qui vous ont poussée à repartir travailler en Italie. Les cinq ans qui ont été nécessaires avant de pouvoir passer à l'essai clinique s'expliquaient-ils par la nécessité d'approfondir certaines connaissances fondamentales ou par des obstacles administratifs ?
M. Philippe Menasché, directeur de l'unité « Thérapie cellulaire en pathologie cardio-vasculaire » à l'hôpital européen Georges Pompidou . Je remercie à mon tour les organisateurs de cette journée de leur invitation. Voilà plus de quinze ans que nous avons commencé de travailler sur la thérapie cellulaire de l'insuffisance cardiaque sans avoir à ce jour obtenu aucun « succès ». Nous avons beaucoup appris, avons conduit des essais chez l'animal et des essais cliniques chez l'homme avec des cellules adultes, et allons prochainement solliciter l'autorisation d'utiliser des cellules cardiaques d'origine embryonnaire - desquelles nous escomptons beaucoup, d'après les observations que nous avons faites.
La France dispose de multiples atouts. Le premier est la qualité scientifique de ses équipes de recherche, trop souvent sous-estimée. En matière de cellules souches embryonnaires humaines, je tiens à citer les travaux de Michel Pucéat, dont la notoriété est aujourd'hui internationale, sans lesquels nous n'aurions pu avancer.
Le deuxième de ces atouts est un vivier dense de PME innovantes en matière de biomatériaux et de dispositifs médicaux.
Le troisième, il faut en rendre hommage à l'AFSSAPS, est l'existence d'une structure, mise en place il y a quelques années maintenant, qui permet aux chercheurs, dès le début d'un projet de thérapie cellulaire, de présenter un projet et, en fonction du retour de l'Agence, de le modifier pour le rendre conforme au cahier des charges et gagner ainsi du temps lors de la soumission définitive.
Un autre atout, assez spécifique à notre pays, réside dans l'absence de cloisonnement entre système hospitalier et système universitaire, qui doit être à tout prix préservée. La plupart d'entre nous sommes des praticiens cliniciens qui avons une activité de laboratoire et nos allers-retours permanents entre la clinique et la recherche nous sont enviés. Les chercheurs d'un très prestigieux institut de recherche à Boston publient énormément sur les cellules souches embryonnaires dans les plus grandes revues internationales comme Science ou Nature . Mais ils sont totalement coupés des applications cliniques.
Enfin, et c'était vrai déjà avant le grand emprunt, quand les projets sont correctement présentés, on finit toujours par trouver les financements nécessaires, même si cela prend du temps.
Mais à côté de ces atouts, il existe des freins. Techniques tout d'abord. Il ne faut pas les sous-estimer car ils peuvent à eux seuls empêcher d'avancer. En pharmacologie, le paradigme est assez simple avec le couple constitué par le récepteur et son ligand. C'est beaucoup plus compliqué en thérapie cellulaire, les contours des produits étant infiniment plus flous.
La première difficulté est de caractériser les produits de thérapie cellulaire. On nous demande pourquoi nous ne réinjectons pas aux patients des cellules souches cardiaques mises en culture après leur avoir été prélevées par biopsie ou au détour d'une intervention chirurgicale. Cette technique ne poserait aucun problème éthique par rapport à l'utilisation de cellules souches embryonnaires. Il est, hélas, très difficile de caractériser une cellule souche cardiaque : plus d'une dizaine de marqueurs ont été décrits, dont la plupart s'excluent mutuellement.
Deuxième difficulté : l'amplification des cellules. Pour « régénérer » - le terme est à la mode, bien qu'on n'ait jamais rien « régénéré » du tout ! - un myocarde infarci, deux milliards de cardiomyocytes sont nécessaires, ce qui est considérable et, je pense, hors de portée. Cela étant, une « régénération » partielle suffit. Mais il existe quand même un effet d'échelle et amplifier des micro-colonies de cellules souches embryonnaires humaines, de surcroît dans des conditions GMP (Good Manufacturing Practice) 2 , constitue un véritable défi technique. Des mois de travail sont nécessaires car il est difficile de maîtriser la prolifération de ces cellules, tout en les maintenant à un stade d'indifférenciation.
Sachant la difficulté de travailler avec des cellules allogéniques, on nous demande pourquoi nous ne travaillerions pas en autologue. Je l'ai fait. Lorsque j'ai effectué les premières greffes de cellules musculaires, je les prélevais dans la cuisse des patients. Cela ne résout hélas pas tous les problèmes. La difficulté de l'utilisation des cellules autologues réside dans la non-reproductibilité et la fonctionnalité variable des cellules d'un patient à l'autre, sans même parler des coûts dus à l'individualisation des contrôles-qualité nécessaires sur chaque lot.
D'où le recours à des cellules allogéniques en provenance de biobanques qui présentent l'avantage, elles, d'être parfaitement caractérisées, mais l'inconvénient d'être susceptibles de rejet, phénomène difficile à contrôler. C'est l'un des freins techniques à lever en priorité.
Une autre difficulté est de maintenir en vie les cellules injectées. Il ne reste pas grand-chose au bout de quelques semaines des injections intra-cardiaques, ce qui naturellement limite l'effet thérapeutique - c'est comme si on ne parvenait pas à garantir que la bio-disponibilité d'un médicament administré à un malade lui permettra d'être actif. Tant que ces problèmes techniques n'auront pas été résolus, on ne peut pas espérer de bénéfice thérapeutique. Et cela n'est pas vrai seulement en cardiologie. Des îlots de Langerhans greffés chez des patients diabétiques meurent eux aussi rapidement.
Il ne faut pas occulter tous ces freins techniques, comme les médias ont parfois tendance à le faire au profit du débat éthique ou sociétal.
Il y a aussi des freins expérimentaux. Nous avons travaillé sept ans sur les cellules musculaires avant notre premier essai clinique et voilà sept ans maintenant que nous travaillons sur les cellules souches embryonnaires, alors que nous allons seulement déposer notre demande d'autorisation d'essai. On nous demande des arguments précliniques. C'est normal, il faut bien entendu travailler en laboratoire - à condition d'avoir toujours présentes à l'esprit les limites de ce travail. Des chercheurs ont démontré qu'une étude expérimentale qui n'a été ni randomisée ni conduite en double aveugle, a quand même une très forte probabilité de déboucher sur un résultat positif. Or, dans la littérature sur la thérapie cellulaire, rares sont les études qui satisfont à ces deux critères rigoureux de randomisation et d'évaluation à l'aveugle.
Les modèles animaux aussi ont leurs limites. L'équipe de Marina Cavazzana-Calvo, elle nous l'a dit, ne pouvait absolument pas prévoir, à partir des essais précliniques réalisés chez la souris, que certains des enfants traités développeraient une leucémie. Nous avons travaillé pendant sept ans sur les cellules musculaires chez des petits animaux, puis de plus gros, sans jamais observer les troubles du rythme ventriculaire qui se sont manifestés chez le troisième malade que j'ai opéré. La plupart des études précliniques sont menées sur de petits animaux. Chacun peut comprendre que lorsqu'on passe à des coeurs beaucoup plus volumineux, les résultats peuvent être très différents. Le coeur de la souris bat à 300/400 pulsations par minute, celui du rat à 300 par minute et celui de l'homme à 60/80 seulement. Lorsqu'on injecte à une souris des cellules embryonnaires humaines, programmées pour battre à un rythme de coeur d'homme, non seulement on se trouve dans le cadre d'une xénogreffe mais de surcroît dans une situation totalement anti-physiologique chez cet animal. Il est alors difficile de tirer des conclusions. La seule façon de sortir de l'impasse de ces essais est de greffer à des singes des cellules embryonnaires de singe, ce que nous avons fait. Mais vous imaginez aisément la complexité logistique et financière de ce type d'étude.

Présentation de M. Philippe Menasché
Outre ces freins expérimentaux, il y a également des freins réglementaires. Dans la phase préclinique, ce sont les contraintes de l'expérimentation animale. Au risque d'être politiquement incorrect, force m'est d'avouer qu'on a presque atteint le délire. Je suis le premier à dire qu'il faut respecter les animaux mais on arrive aujourd'hui à un coût de 170 euros par mètre carré pour héberger et conditionner certaines souris dans des conditions répondant à toutes les exigences réglementaires - soit dix fois plus que le prix des loyers payés par certains de nos concitoyens !
Le poids de l'évaluation constitue un autre frein. Le système actuel s'apparente à un millefeuille dans lequel il n'est pas facile de se retrouver. Nous finissons pas passer plus de temps à nous évaluer et à évaluer les autres qu'à travailler. Là aussi, il faudrait plus de mesure.
Il existe aussi des freins réglementaires de nature clinique. En matière de thérapie cellulaire, il n'y a actuellement aucune homogénéité en Europe même si l'EMA ( European Medicine Agency ) essaie d'uniformiser les pratiques. Les contraintes demeurent différentes selon les pays, ce qui ne facilite pas la tâche dans le cas d'essais multicentriques. Je ne reviens pas sur la lourdeur des essais cliniques, ni sur les contraintes qui les entourent.
Enfin, le principe de précaution pèse également. Les patients gravement atteints sont en général partants pour des thérapeutiques nouvelles, dès lors qu'on leur a correctement expliqué les enjeux et les risques. Mais il faut composer avec la réglementation. Aujourd'hui, il serait absolument impossible, avec l'encadrement qui est le nôtre, de réaliser une première transplantation cardiaque !
Je voudrais vous présenter en conclusion un classement, effectué il y a quelques années, de plusieurs pays, en fonction de critères à la fois médicaux, scientifiques et industriels. Sur une échelle de 0 à 5, arrive en premier le Royaume-Uni qui a une réglementation rigoureuse mais tout de même plus permissive que la nôtre - le pays dispose de très bonnes équipes médicales, d'un très bon niveau scientifique et maintenant de nombreuses entreprises de biotechnologies. Les Etats-Unis se classent de façon différente - il est d'ailleurs difficile de les classer, la législation ayant changé avec l'administration Obama : ce qui y prime, c'est l'implication des entreprises de biotechnologies et même des grandes firmes pharmaceutiques en matière de thérapie cellulaire. La Chine et la Corée sont à peu près à égalité : la réglementation y est très lâche, la recherche clinique assez peu développée et la recherche scientifique théorique, de bon niveau, en amélioration constante, notamment en Chine. La France, elle, a une position très équilibrée, qui reflète assez bien l'équilibre entre un véritable esprit d'innovation, lié à la fois à nos équipes de chercheurs et à notre expertise en recherche translationnelle - mise en application médicale des résultats scientifiques de la recherche fondamentale - et en recherche clinique, et les freins qui nous sont imposés. Nous comprenons ce qui peut les motiver, mais il serait souhaitable que soit parfois mieux prise en compte l'évaluation du rapport bénéfice/risque.
M. Luc Sensebé, directeur médical et scientifique de l'Établissement français du sang (EFS) de la région Centre . Je vous prie d'avance de m'excuser si je suis conduit à répéter certains points déjà évoqués par Marina Cavazzana-Calvo et Philippe Menasché. Je voudrais m'efforcer de montrer que certains échecs peuvent permettre de « rebondir » et conduire in fine à des découvertes intéressantes. Mon exposé portera sur les cellules souches mésenchymateuses et la maladie du greffon contre l'hôte (GVHD) - les cellules immunitaires du greffon reconnaissent l'hôte comme étranger et attaque certains de ses tissus.
Les cellules souches mésenchymateuses, outre la capacité à se différencier dans la totalité des tissus squelettiques, mais aussi en tissu musculaire ou vasculaire et, dans certaines conditions, en d'autres tissus, agissent au cours de la réparation tissulaire grâce à la production de facteurs de croissance et de facteurs trophiques, et ont une action à la fois immuno-modulatrice et anti-inflammatoire. Elles sont capables d'agir sur toutes les cellules de l'immunité, ce qui en fait un outil intéressant en thérapie cellulaire, pour aider à la solution de certains problèmes lors de greffes ainsi que dans le cas de pathologies inflammatoires ou immunitaires.
La GVHD aiguë est un risque majeur lors de toute greffe de moelle, le risque de décès oscillant de 15% à 20%. Le premier succès intervenu dans le traitement de cette maladie par des cellules souches mésenchymateuses a été rapporté en 2004 par l'équipe de Katarina Le Blanc au Karolinska Institutet de Stockholm. Les recherches avaient commencé dès 1990 mais les applications cliniques ont attendu 2004. Depuis lors, les cellules souches mésenchymateuses ont été utilisées chez plusieurs centaines de patients pour traiter une GVHD résistante au traitement classique. Dans cette situation difficile, on obtient de bons résultats en particulier pour traiter les atteintes digestives. Les résultats obtenus sont par ailleurs meilleurs chez l'enfant que chez l'adulte.
S'appuyant sur les connaissances acquises, la Société française de greffe de moelle et de thérapie cellulaire (SFGM-TC), qui réunit l'ensemble des greffeurs de cellules souches hématopoïétiques 3 , a travaillé pendant deux ans, avec l'aval des autorités de régulation, à l'élaboration d'un protocole de production satisfaisant aux exigences à la fois réglementaires et de sécurité sanitaire pour leur utilisation en clinique chez l'homme. Après mise en place d'un procédé de production standardisé, la SFGM-TC a obtenu un financement du Programme hospitalier de recherche clinique, le PHRC, pour l'élaboration d'un protocole de prévention de la GVHD par injection de cellules souches mésenchymateuses. Ce protocole a mobilisé 21 centres de greffe en France. L'AFSSAPS a donné son autorisation fin 2006.
Les inclusions ont débuté en février 2007, avec un objectif de 78 patients. Cinq mois plus tard, en juillet, après l'inclusion de 11 patients, on a constaté dans deux cas une anomalie du caryotype des cellules cultivées. Bien qu'il n'y ait eu chez les patients aucun incident grave en rapport avec cette anomalie, le protocole a été suspendu et l'incident immédiatement déclaré à l'AFSSAPS. Dès cette suspension, les équipes ont recherché les raisons de ces anomalies et analysé leurs conséquences potentielles. Au terme de ces études complémentaires, qui ont pris en gros neuf mois, nous avons démontré que les anomalies caryotypiques susceptibles d'advenir en culture n'étaient pas récurrentes, qu'il n'y avait pas d'avantage sélectif - les cellules anormales ne prennent pas le pas sur les autres - ni de transformation des cellules pouvant aboutir à la greffe de cellules anormales, et au total qu'il n'y avait donc pas de risque supplémentaire pour les patients. Ces résultats ont été présentés en congrès international dès 2008, puis publiés. L'AFSSAPS n'a toutefois levé la suspension de l'essai que deux ans plus tard, au printemps 2010, et demandé que les cellules souches mésenchymateuses ne soient plus utilisées qu'en curatif et non en préventif.
Notre échec, qui n'était pas prévisible et nous a obligé à revoir nos méthodes, aura finalement été positif. On comprend désormais mieux les anomalies liées à la culture, ce à quoi personne ne s'était intéressé jusqu'alors. On a développé des contrôles spécifiques avec des cibles différentes pour contrôler la sécurité de ces cellules. Et au bout du compte, un nouveau procédé de culture a été mis au point, désormais largement utilisé - divers protocoles en cardiologie et en neurologie y ont actuellement recours. Il a enfin servi de base pour des protocoles dans divers essais européens, subventionnés par l'Union européenne, dans deux consortiums coordonnés par la France, en particulier par l'Établissement français du sang et l'INSERM, dans le domaine de l'orthopédie et des maladies de la peau et de la cornée.
Quelles sont les conditions de la réussite ? Il faut tout d'abord une très grande réactivité de tous les acteurs - recherche, recherche-développement, clinique et autorités réglementaires - en dépit de la complexité. Il faut également des liens forts entre recherche cognitive et recherche-développement et une étroite collaboration entre les équipes des deux secteurs. Il faut aussi mieux évaluer tous les aspects du rapport bénéfice/risque : les bénéfices sont aujourd'hui minimisés. Il faut éviter les évaluations répétitives : sans nouvel élément scientifique ou clinique, dès lors qu'une instance compétente et reconnue a procédé à une évaluation, c'est celle-ci qui doit faire foi. Enfin, dans la mesure où l'administration de produits de thérapie cellulaire chez l'homme ne sera autorisée que si les essais de phase 1 et 2 - sécurité puis efficacité - sont reproductibles et acceptés par les agences de régulation, en particulier l'Agence européenne des médicaments, il faut respecter les conditions relatives aux GMP ( Good Manufacturing Practices ) 4 dès les phases 1et 2. Sinon on risque d'être bloqué pour aller au-delà.
Pour ce qui est des financements, je suis globalement d'accord avec Philippe Menasché, il existe de l'argent pour la recherche. Il n'est pas trop difficile de financer les essais cliniques. Il l'est beaucoup plus de financer l'étape, très coûteuse, qui permet de passer de la recherche à la fabrication d'un médicament utilisable chez l'homme. Le passage aux essais cliniques de phase 3 et 4 est très long.
Mme Emmanuelle Prada-Bordenave . Vos recherches ont été bloquées pendant deux ans. Alors que vous aviez eu l'impression d'apporter toutes les justifications en 2008, ce n'est qu'en 2010 que vous aviez été autorisé à reprendre vos recherches. Mais grâce à la réactivité de votre équipe, ce retard pour certains patients en hématologie a finalement profité à d'autres puisque durant cette période-là, vous avez pu affiner votre méthode, qui est devenue du coup utilisable dans d'autres champs de la médecine régénérative.
M. Luc Douay, directeur médical et scientifique de l'Établissement français du sang (EFS) d'Île-de-France . Chaque année, 90 millions de culots globulaires, qu'on appelle plus couramment poches de sang, sont transfusés de par le monde, dont un tiers en Europe, en Amérique du Nord et au Japon. En France, on transfuse chaque année 2,5 millions de ces culots, grâce à 1,7 millions de donneurs bénévoles qui effectuent en moyenne 1,6 don par an - il en faudrait deux pour couvrir en permanence la demande, qui est en augmentation régulière.
Si dans les pays développés, la préoccupation est d'améliorer sans cesse la qualité des produits sanguins - en effet, plus on transfuse les patients, plus augmente le risque d'allo-immunisation -, de pouvoir fournir des produits sanguins adéquats aux patients de groupes rares et répondre à d'éventuelles situations de crise, dans les pays en voie de développement, un gros problème de santé publique aurait été résolu si on pouvait y fournir en permanence un produit sanguin de qualité et présentant toute sécurité sanitaire.
Pourquoi chercher de nouvelles sources de globules rouges ? La population vieillit et au fil de l'âge, l'incidence des pathologies exigeant des transfusions sanguines augmente. On manquera donc nécessairement de donneurs à un moment car il n'est pas possible de donner son sang au-delà d'un certain âge.
On rêve depuis des décennies d'un sang artificiel. On travaille depuis longtemps à la fabrication de substituts synthétiques, fabriqués à partir d'hémoglobine humaine ou animale. Malheureusement, une méta-analyse conduite en 2008 sur l'ensemble des essais pratiqués dans le monde a montré que les patients ayant reçu de tels substituts, quelle qu'en soit la nature, avaient 30% de risques de plus de décéder d'un infarctus du myocarde. Cela a, temporairement sans doute, conduit à stopper ces travaux.
Avec les progrès des connaissances en matière de cellules souches et de thérapie cellulaire, on s'est naturellement demandé si on ne pourrait pas fabriquer des globules rouges à partir de cultures de cellules souches, qu'on trouve dans la moelle osseuse, dans le sang à condition d'y mobiliser un facteur de croissance particulier, et, on le sait depuis une vingtaine d'années, dans le sang du cordon ombilical - qu'on appelle en France le sang placentaire.

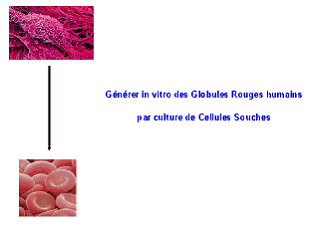
Présentation de M. Luc Douay
Notre équipe a, elle, créé le concept de globule rouge de culture en 2002 et l'a développé surtout depuis 2005, l'idéal étant de parvenir à produire in vitro des poches de sang de phénotype universel à partir d'une source illimitée de cellules souches, et ce de façon industrielle.
La première étape était de passer de la cellule souche au globule rouge. Il nous a fallu reconstituer in vitro le micro-environnement de la moelle osseuse et mettre au point un protocole d'entretien de ces cellules souches - on reconstitue dans un flacon un micro-environnement à partir de cellules souches mésenchymateuses, on y dépose des cellules souches hématopoïétiques et on voit progressivement, en l'espace de quelques semaines, naître des globules rouges. Je ne détaille pas toutes les étapes de la maturation du globule rouge : mature, celui-ci a la forme d'un disque déformable, ce qui lui permet de circuler partout dans les capillaires.
La deuxième étape était de démontrer que les globules rouges ainsi obtenus étaient fonctionnels. Nous avons prouvé in vitro que leur hémoglobine était capable de transporter l'oxygène. Nous les avons aussi injectés à des souris transgéniques chez qui leur maturation s'est révélée tout à fait normale.
Mais sachant que dans chaque poche de sang transfusée, il y a 2 000 milliards de globules rouges, le défi est d'atteindre en laboratoire ce niveau de production.
En couplant les connaissances acquises en matière de greffe de moelle osseuse, de cellules souches hématopoïétiques et de facteurs de croissance de l'hématopoïèse, on peut combiner une première phase d'amplification des cellules souches à une deuxième phase de différenciation en globule rouge. A partir d'une cellule souche de sang de cordon, on peut générer plus de 40 millions de globules rouges, ce qui signifie qu'en partant d'une unité de sang placentaire, soit 80 à 100 ml, on peut produire l'équivalent de 75 poches de sang.
Disposer d'une source illimitée de cellules souches permettrait de s'affranchir des contraintes d'approvisionnement.
Le modèle d'étude, idéal et indispensable, est celui de la cellule souche embryonnaire. Rien de ce que je vais vous présenter maintenant n'aurait été possible sans travail sur les cellules souches embryonnaires. Mais dans notre application, le choix du phénotype des globules rouges étant impossible à partir d'une cellule souche embryonnaire, il fallait trouver une autre solution. Elle est venue des cellules somatiques adultes dédifférenciées puis reprogrammées pour retrouver un état de pluripotence, les fameuses iPS (induced pluripotent stem cells) , découvertes par les équipes japonaise de Shinya Yamanaka et américaine de James Thomson. Ces iPS sont capables de donner naissance à l'ensemble des tissus de l'organisme. Nous avons, pour notre part, montré, qu'il était possible de produire à partir d'elles des globules rouges fonctionnels.
On dispose donc de deux sources de cellules souches pour un même produit final, le sang de cordon et les iPS. Chacune a ses avantages et ses inconvénients. Si avec un sang de cordon, on peut produire des quantités considérables de globules rouges, la ressource est ponctuelle. Une cellule iPS, elle, ne produit que mille globules rouges, mais la ressource est illimitée.
Pour satisfaire l'ensemble des besoins transfusionnels français avec du sang placentaire, il faudrait 50 000 cordons par an. C'est tout à fait concevable dans la mesure où cela ne représente que 6% des 800 000 naissances annuelles et où on sait gérer les banques de sang placentaire. Il y en a des centaines de milliers dans le monde, et une dizaine de milliers pour l'instant en France, où l'objectif est de les développer. Mais il faudrait s'organiser pour collecter du sang de tous les groupes sanguins en sélectionnant les sangs de cordon.
De l'autre côté, une étude a montré qu'en choisissant judicieusement les donneurs de ces cellules souches, nous pourrons disposer d'une source de globules rouges quasi universels. En effet, 3 donneurs seulement, présentant une expression particulière des principales familles de groupes sanguins, permettront de répondre aux spécificités transfusionnelles de plus de 99% des receveurs. On serait très près du donneur universel.
L'intérêt de produire des globules rouges de culture à partir d'iPS est qu'il s'agit d'une source sûre - on peut parfaitement maîtriser cette population cellulaire et éviter toute contamination par des agents pathogènes -, illimitée, capable de fournir le phénotype voulu. Mais l'outil reste à parfaire et tout n'est pas encore réglé. Il faut par exemple sécuriser la reprogrammation afin que ces cellules ne provoquent pas de tumeurs à cause du matériel génétique résiduel. Le globule rouge étant une cellule dépourvue de noyau, le risque s'en trouve considérablement limité. Il faut ensuite améliorer la capacité d'amplification des iPS, contrôler leur différenciation en globule rouge, et surtout définir des conditions de grade clinique de leur production.
Pour ce qui est de la santé publique et du rapport bénéfice/risque, on passerait d'un modèle où un receveur reçoit aujourd'hui des globules rouges de plusieurs donneurs, ce qui, à chaque fois, augmente les risques, théoriques et réels, d'allo-immunisation et de transmission d'agents infectieux, à un modèle où le sang d'un seul donneur pourrait être transfusé à plusieurs receveurs, ce qui ne poserait de problème que si on n'était pas en mesure de garantir la parfaite sécurité du produit, ce qui n'est pas le cas. Avec ces globules rouges de culture, nous répondrions aux besoins particuliers à la fois des pays développés et des pays en développement que j'exposai au début de ma présentation.
L'enjeu est aujourd'hui de passer d'une production artisanale à un développement industriel. Le plus important est de convaincre les industriels, au nom à la fois de la santé publique et de la compétition internationale, de se lancer dans l'aventure. Dans le parcours du combattant qui a été le nôtre pour faire admettre l'intérêt du projet, la première difficulté a été de convaincre de sa faisabilité. Nous y sommes parvenus. Une autre a été de trouver des financements : la première partie du projet a été financée par de petites associations privées, ce qui nous a permis de publier nos travaux. Le relais a été pris, il faut s'en réjouir, par l'Établissement français du sang qui, considérant que ce projet participait de son objectif d'améliorer toujours la qualité des produits sanguins, nous a beaucoup aidés.


Présentation de M. Luc Douay
Mais ce qui a littéralement « boosté » nos recherches, c'est que l'armée américaine, par le biais de son organisme de recherche, la DARPA ( Defence Advanced Research Projects Agency) , s'y est intéressée, jugeant que c'était un moyen de pouvoir approvisionner ses troupes en produits sanguins partout dans le monde 24 heures sur 24. Elle a lancé un programme Blood Pharming afin de produire des globules rouges à partir du sang de cordon, n'hésitant pas à imaginer un appareil tenant dans un mètre cube et pouvant fabriquer des globules rouges sur le théâtre même des opérations. Il a fallu ce détour par l' US Army pour valider notre approche en France.
Les Britanniques, eux, travaillent plutôt à partir des cellules souches embryonnaires, sur lesquelles je vous ai fait part de mes réserves.
En France, nous avons récemment monté le projet « StemRed » grâce à OSEO, en partenariat avec l'Établissement français du sang, l'université Pierre et Marie Curie et plusieurs entreprises de biotechnologies. Il est désormais opérationnel.
Où en est-on de l'application clinique ? Les premiers essais sont en cours à l'hôpital Saint-Antoine à Paris. Ils sont réalisés à partir de cellules souches périphériques d'un donneur intra-familial dont le membre de fratrie recevra une greffe de cellules souches hématopoïétiques. Les globules rouges fabriqués sont marqués pour pouvoir être suivis in vivo .
Nous saurons d'ici trois ans, c'est notre objectif, s'il est possible de passer aux essais cliniques de stade supérieur. Là, d'autres obstacles devront être franchis.
Mme Emmanuelle Prada-Bordenave . Je remercie les quatre intervenants pour leurs remarquables exposés. Tous ont fait preuve d'une grande honnêteté dans la présentation des difficultés auxquelles ils ont pu se heurter, parfois inattendues, si surprenantes qu'elles auraient pu amener certains - peut-être cela a-t-il d'ailleurs été le cas - à céder à la peur face à l'inconnu, ce qui n'est pas la bonne réaction. Il appartient aux autorités sanitaires, mais aussi politiques, notamment à la représentation nationale, et je remercie le président Birraux d'avoir pris l'initiative d'une rencontre comme celle d'aujourd'hui, de réfléchir aux moyens de surmonter ces peurs irrationnelles en trouvant des solutions qui préservent la sécurité sanitaire et l'intérêt collectif, tout en permettant à la science d'avancer. J'ouvre maintenant le débat.
DÉBAT
M. le président Claude Birraux . Vous nous avez, Madame Cavazzana-Calvo, laissés sur notre faim. Pourriez-vous nous dire précisément pourquoi vous n'avez pas pu continuer vos travaux en France et êtes repartie en Italie ?
Mme Marina Cavazzana-Calvo . La France m'a donné à la fin des années 90 la possibilité de mener des recherches uniques. Tout s'est, hélas, ensuite arrêté. On ne m'a pas permis de développer un laboratoire de recherche à la hauteur des possibilités en matière d'essais cliniques. Lasse de toutes ces difficultés, j'ai « exporté » en Italie les projets que je ne pouvais plus conduire ici. J'ai pris l'an dernier un congé sabbatique pendant lequel j'ai monté deux projets cliniques en Italie. En trois mois seulement, j'ai pu constituer là-bas, avec une structure pourtant moins performante qu'ici, deux équipes de dix personnes travaillant l'une sur un projet de thérapie génique du sida, l'autre sur l'utilisation thérapeutique de cellules amniocytaires - projet auquel j'avais commencé à travailler il y a quelques années en France avec le professeur Frydman et que je n'ai pu poursuivre faute de moyens financiers, humains et logistiques. Nous allons bientôt publier. C'est clairement parce que je ne dispose pas des moyens nécessaires en France que je suis allée en Italie développer ces projets, mais je travaille encore en France.
M. Olivier Boisteau, président des Laboratoires Clean Cells . Je suis membre du comité Biotechnologies du LEEM (Les Entreprises du Médicament), organisation qui regroupe les entreprises de l'industrie pharmaceutique, et président d'un laboratoire qui fabrique des produits de thérapie cellulaire. Je puis à ce double titre affirmer que des travaux d'une qualité exceptionnelle sont menés en France depuis longtemps et que des avancées réelles ont été obtenues. Il existe certes aujourd'hui des freins scientifiques et techniques mais aussi réglementaires, qu'il faudrait lever car certains pays, loin de disposer de nos compétences scientifiques, progressent néanmoins plus vite que nous. Notre pays perd l'avance qu'il avait en matière de thérapie cellulaire - dont l'AFSSAPS avait pris la mesure puisqu'elle avait délivré les autorisations nécessaires. Il faudrait aussi harmoniser les réglementations entre pays européens, sachant que l'Agence européenne du médicament, l'EMA (European Medicine Agency) , impose désormais des conditions GMP ( Good Manufacturing Practice ).
Le régime d'interdiction des recherches sur les cellules souches embryonnaires - assortie d'un moratoire qui arrive d'ailleurs à expiration le 5 février 2011 - actuellement en vigueur en France, constitue un frein. Ces cellules en effet peuvent être utiles non seulement en thérapeutique mais aussi pour tester la toxicité des substances pharmacologiques, ce qui permettrait de mieux garantir la sécurité des médicaments. Les iPS constituent elles aussi un remarquable outil de criblage moléculaire. Le principe de précaution l'emporte hélas souvent sur une évaluation objective de la balance bénéfices/risques et pousse à se focaliser sur les seconds. Les iPS permettront d'établir de manière encore plus efficace qu'aujourd'hui la toxicité ou l'innocuité des médicaments, parmi lesquels les produits de thérapie cellulaire, mais aussi les anticorps monoclonaux et autres protéines recombinantes. Il faut que la nouvelle loi de bioéthique autorise la recherche sur les cellules souches embryonnaires tout en l'encadrant, au lieu de continuer de l'interdire en accordant des dérogations au cas par cas.
Il serait dommage que la France perde son avance en matière de thérapie cellulaire et génique à cause d'une application excessive du principe de précaution, d'une évaluation timorée du bénéfice des médicaments et de lois de bioéthique qui, au final, nous desservent. En Chine, aux Etats-Unis ou au Royaume-Uni, chercheurs et industriels ne sont pas soumis aux mêmes contraintes. Ces pays, qui étaient en retard sur nous, nous rattrapent et nous devanceront si nous ne faisons rien.
M. le président Claude Birraux . Le projet de loi relatif à la bioéthique, qui révise la loi de 2004, est en cours d'examen au Parlement.
Il faut, me semble-t-il, être prudent. Lors d'une réunion européenne des instances parlementaires homologues de l'OPECST, un représentant britannique se désolait que certains politiques invoquent l'éthique là où il aurait fallu, selon lui, parler business, et empêchent de gagner tout le « fric » qu'il serait possible de gagner. Je ne peux accepter une telle approche. Il ne suffit pas de réfléchir à ce qui est possible, il faut aussi réfléchir à ce qui est acceptable. Nous connaissons mal la Chine mais je ne suis pas sûr que l'éthique y soit l'une des préoccupations principales...
M. Jean Marimbert, directeur général de l'Agence française de sécurité sanitaire des produits de santé (AFSSAPS) . En tant que directeur général de l'AFSSAPS, j'ai eu à connaître ces dernières années de projets porteurs de progrès thérapeutiques potentiels qui ne posaient de problèmes ni sur le plan scientifique ni pour l'évaluation de la balance bénéfices/risques ni sur le plan éthique, mais où l'obstacle est venu d'une certaine interprétation de la loi - chacun voit à quoi je fais allusion. Après des négociations extrêmement difficiles avec de hautes autorités il y a un an et demi, nous avons obtenu que Philippe Menasché puisse soumettre la demande d'essai clinique qu'il va prochainement déposer à l'Agence de la biomédecine, au bénéfice d'une interprétation qui était tout à fait possible de la loi. Nous avons rencontré des difficultés analogues pour la vitrification ovocytaire. Mme Prada-Bordenave et moi-même avions, dans les deux cas, le sentiment d'avoir affaire à des projets tout à fait valides sur le plan scientifique, susceptibles de passer avec succès le cap de l'évaluation qui, de toute façon, demeurera nécessaire, quelque articulation que l'on conçoive entre éthique et principe de précaution, et ne soulevant pas de problèmes éthiques majeurs. Philippe Menasché souhaitait ainsi utiliser des cellules souches embryonnaires humaines après plusieurs essais cliniques avec des cellules souches adultes qui s'étaient, hélas, tous soldés par un échec. Les conditions dans lesquelles une dérogation peut être accordée, notamment celle « d'absence de méthode alternative d'efficacité comparable », étaient parfaitement respectées. Quant à la technique innovante de vitrification des ovocytes, elle était même susceptible de réduire le problème éthique en permettant de limiter le nombre d'embryons surnuméraires. Si les agences sanitaires peuvent être à l'origine de retards, encore que leur accompagnement ait souvent été moteur comme pour les essais cliniques évoqués par Mme Cavazzana-Calvo, elles se heurtent elles-mêmes parfois à des obstacles tenant à une interprétation restrictive de l'état du droit. J'attends beaucoup sur ce point du futur projet de loi relatif à la bioéthique.
Mme Emmanuelle Prada-Bordenave . Madame, messieurs, avez-vous rencontré des difficultés pour nouer des partenariats avec de petites entreprises de biotechnologies, faisant par exemple de la sécurisation virale ou susceptibles de vous aider à passer en conditions GMP ( Good Manufacturing Practice ), toutes choses que ne font pas les établissements hospitaliers ni les laboratoires de recherche académiques ? Y a-t-il assez d'entreprises de ce type en France ? Êtes-vous obligés de faire appel à des entreprises étrangères, comme c'est actuellement le cas pour les cultures de peau destinées aux grands brûlés ? Ces petites entreprises se heurtent-elles à des obstacles administratifs ou financiers ?
M. Luc Douay . Certaines petites entreprises nationales peuvent nous aider ponctuellement. Mais, je l'ai dit, lorsqu'il s'est agi de développer un outil industriel de grande ampleur pour produire des globules rouges de culture, ce n'est pas en France que nous avons noté le plus d'enthousiasme. C'est l' US Army qui a sollicité les entreprises de biotechnologies américaines qui se sont les premières engagées. Ce n'est qu'ensuite que nous avons pu convaincre des entreprises françaises de se lancer. Elles étaient au départ très frileuses.
M. Jean-Sébastien Vialatte, député du Var, membre de l'OPECST . Lors de l'examen du projet de loi relatif à la bioéthique qui s'est achevé la nuit dernière en commission, plusieurs de nos collègues ont exprimé une très vive méfiance à l'égard de l'industrie pharmaceutique s'agissant de l'utilisation des cellules souches embryonnaires, craignant notamment une dérive marchande de l'utilisation des produits du corps humain. A un degré moindre, transparaissait dans leurs propos une crainte vis-à-vis de la recherche. Si le Parlement devait aujourd'hui adresser un message, il faudrait être très prudent sur les promesses thérapeutiques, notamment leur horizon, car le public, qui ne mesure pas nécessairement le temps nécessaire au développement des recherches et des essais cliniques, déçu, en vient du coup à se défier de la recherche et des chercheurs.
M. Philippe Menasché . Pour notre part, nous avons toujours trouvé en France les entreprises de biotechnologies dont nous avions besoin pour certaines prestations de service, qu'il s'agisse de sécurisation virale ou de fabrication de biomatériaux. Récemment encore, j'ai été émerveillé des technologies d'entreprises de Besançon et sa région dont nous avons sollicité l'expertise pour réaliser des tubes destinés à la reconstruction de la voie pulmonaire chez des enfants souffrant de cardiopathies congénitales. Pour ce qui est de la culture de peau, la société étrangère à laquelle vous avez fait allusion a commencé d'y travailler il y a quinze ou vingt ans, ce qui explique aujourd'hui son avance et sa notoriété. Nous avons en France un vivier dense de petites entreprises de biotechnologies très motivées. Il est vrai que les choses changent lorsqu'on arrive aux essais de phase 3 ou 4. Mais là, ce sont les grandes firmes pharmaceutiques qui sont concernées. Leur attitude est en train de changer, et elles regardent dorénavant de près les débouchés que pourrait leur offrir la thérapie cellulaire.
Mme Marina Cavazzana-Calvo . Je suis plutôt d'accord avec M. Douay. Il n'y a pas en France d'entreprises de biotechnologies qui soutiennent de façon importante la thérapie génique, à l'exception du Généthon, créé en 1990 à partir des dons collectés lors du Téléthon.
S'agissant de la recherche sur les cellules souches embryonnaires, il est extrêmement important d'expliquer qu'elle n'a pas que des finalités thérapeutiques. La rédaction de la dernière loi de bioéthique constituait à cet égard une erreur. La recherche sur ces cellules est un outil-clé pour comprendre le développement du corps humain et les maladies rares. Continuer de ne l'autoriser qu'à des fins thérapeutiques accentuerait encore le retard, déjà colossal, de la France, que j'évalue à dix ou quinze ans. Quant au maintien du régime dérogatoire, il ferait prendre dix ans de retard supplémentaires.
M. Jean de Kervasdoué, ancien directeur général des hôpitaux, membre de l'Académie des technologies, professeur au Conservatoire national des arts et métiers . Philippe Menasché a évoqué la très grande complexité en France de la gestion administrative des projets de recherche, parlant de « millefeuille ». Que les universités doivent obtenir des autorisations pour quantité de recherches est source de retards considérables.
La situation est un peu meilleure pour la création d'entreprises grâce à la législation favorisant le développement du capital-risque. Nous avons néanmoins un énorme retard, dont je doute que nous le rattrapions. Comme l'a expliqué Luc Douay, c'est seulement après que l' US Army s'est déclarée intéressée par un projet et y a pris part, que certaines entreprises françaises ont accepté d'investir dans le même champ. Je pourrais vous citer d'autres exemples où l'armée américaine a permis que des recherches se développent en France.
Un pays qui a interdit les recherches sur les cellules souches embryonnaires, sauf dérogation, qui a institué un moratoire sur les OGM et commence d'être on ne peut plus réservé sur les nanotechnologies tout en affirmant que la recherche et l'industrie constituent son avenir, devrait s'interroger.
Nous avons la chance d'avoir encore une industrie pharmaceutique en France - de fait, nous n'avons qu'une entreprise de taille mondiale, dont notre pays ne représente d'ailleurs que 6% des marchés. Nous sommes engagés dans une compétition mondiale. Bien entendu, il n'est pas question de renier nos valeurs éthiques, mais il faut reconnaître que nous sommes entrés, comme l'Allemagne d'ailleurs, dans un délire, à savoir le principe de précaution, particulièrement absurde. Ce principe veut en effet qu'en cas d'événement incertain, il faille prendre des mesures proportionnées. Or, quiconque a un minimum de logique sait qu'incertain et proportionné sont contradictoires. Lors de l'épidémie de grippe H1N1, notre pays, pris de folie, a commandé 10% des vaccins mondiaux et 30% du Tamiflu mondial, alors qu'il ne représente que 1% de la population mondiale. Il a aussi mis en place un dispositif spécifique de vaccination, dont ont été exclus les médecins généralistes. Pour quel résultat ? On a vacciné moins que les médecins traitants ne le font d'ordinaire pour la grippe saisonnière et de toute façon, le virus H1N1 était arrivé avant les vaccins ! Au total, la France n'a pas fait mieux que la Pologne qui, elle, n'a rien fait du tout.
Nous ne sommes pas sur la bonne voie dès lors que des tribunaux reconnaissent le préjudice d'anxiété causé par les antennes relais de téléphonie mobile, alors qu'on le sait, l'énergie qu'elles diffusent est dix mille fois inférieure à celle émise par les antennes de télévision ou la lumière du jour. On pourrait conseiller à ceux qui ont peur de ces antennes de ne sortir aussi que la nuit pour se garder de la lumière du jour !
Nous ne sommes pas non plus sur la bonne voie lorsqu'on tolère que M. Bové et ses amis fauchent des champs de cultures OGM, pourtant ensemencés pour tester certaines de leurs propres hypothèses - c'était le cas de l'expérience conduite par l'INRA sur la vigne en Alsace. La messe est dite concernant les OGM. Treize millions d'agriculteurs dans le monde en produisent, un milliard d'hommes en consomment et les porcs bretons sont nourris d'OGM américains. En cette affaire, la France a tout perdu. L'INRA est bloqué dans ses recherches sur le sujet, Limagrain a perdu son avance dans le domaine des biotechnologies, et nous importons des OGM.
Au-delà des craintes de nos concitoyens, le Parlement doit se saisir de ces questions. Notre pays possède des atouts uniques et sa recherche est encore remarquable. Mais chaque fois qu'un doctorant part à l'étranger, c'est un chèque de 300 000 euros que la France fait à son pays d'accueil - c'est le prix de toute l'excellente formation antérieure dispensée à un doctorant. Si les chercheurs ne disposent pas de l'autonomie nécessaire pour développer leurs recherches en France, ils partiront. Le Parlement doit réagir.
Des raisons religieuses avaient été invoquées pour interdire les recherches sur les cellules souches embryonnaires. Nous étions allés à plusieurs, dont le professeur Frydman, demander au Président Chirac que soit levé le moratoire. Il nous a fort aimablement reçus mais aucune décision n'a suivi. Et les équipes de recherche quittent notre pays et n'y reviendront pas. Nous sommes profondément attachés à certaines valeurs éthiques qu'il n'est pas question de renier mais cette éthique ne doit pas conduire de facto à la paralysie, à moins qu'on ne la souhaite. Mais en ce cas, il faudrait avoir le courage de le dire.
M. le président Claude Birraux . S'agissant des antennes relais de téléphonie mobile, plusieurs tribunaux et cours d'appels sont revenus sur la jurisprudence de la cour d'appel de Versailles qui avait fait une application, contestée et contestable, du principe de précaution.
L'OPECST a organisé une audition, ouverte à la presse, sur le principe de précaution, quatre ans après sa mise en oeuvre. J'ai encore en mémoire ce que nous disait Mme Christine Noiville sur la jurisprudence extrêmement prudente de la Cour de justice des communautés européennes. Il existe une vision dynamique du principe de précaution en tant que principe d'action. Si nos concitoyens sont devenus méfiants et si le débat sur les nanotechnologies a tourné comme il l'a fait, n'oublions pas que c'est parce qu'un chercheur a déclaré un jour qu'il aimerait bien implanter quatre nanoparticules dans quatre cerveaux humains et les relier à un micro-ordinateur, ce qui a nourri tous les fantasmes. Il faut raison garder, de part et d'autre.
M. Jean de Kervasdoué . Dans mon récent ouvrage La peur est au-dessus de nos moyens, je passe en revue la jurisprudence française et européenne. Le principe de précaution de la Charte de Rio n'est pas le même que celui en vigueur dans certains pays européens, lequel n'est lui-même pas identique à celui que nous avons inscrit dans notre bloc de constitutionnalité. Le principe de précaution, qui relevait du droit public est en train de basculer dans le champ du droit privé et alors qu'il ne concernait initialement que l'environnement, il touche maintenant la santé. L'évaluation à laquelle vous avez procédé date tout de même d'un certain temps. Le sujet demeure d'actualité.
M. le président Claude Birraux . Nous en avons traité il y a deux ans. Et mon avis sur le sujet n'a pas changé depuis.
M. Jean de Kervasdoué . Breton moi aussi, je sais qu'un Breton ne change pas facilement d'avis ! Je dis simplement que ce principe a modifié la pratique médicale et que les médecins se tiennent aujourd'hui de plus en plus sur la défensive, par peur des recours en justice. La conjonction des dispositions de la loi Kouchner relative aux droits des malades et l'application du principe de précaution conduisent d'ailleurs à une très grande brutalité envers les malades, auxquels on est obligé, pour être couvert, d'exposer de façon détaillée tous les risques qu'ils encourent et de communiquer même les pourcentages de survie et de décès. Il faut avoir conscience des sommes que nous dépensons vraisemblablement pour rien. D'après une équipe de l'Institut Pasteur, le dépistage de certains virus dans les dons de sang coûte 850 millions d'euros l'année de vie sauvée, et cela au nom du seul principe de précaution.
M. le président Claude Birraux . Et l'affaire du sang contaminé ? On ne peut pas oublier que des patients sont morts d'une infection par le VIH après une transfusion.
M. Jean de Kervasdoué . En l'espèce, le principe de précaution n'aurait servi à rien.
M. le président Claude Birraux . Si, puisqu'on savait qu'on pouvait chauffer les poches de sang et qu'on ne l'a pas fait.
M. Jean de Kervasdoué . J'étais directeur des hôpitaux à l'époque. Je connais bien cette affaire, qui comporte en réalité trois volets. Tout d'abord, le non-rejet des dons à risque - c'est pour cela qu'en France tant de personnes transfusées ont été contaminées -, puis la rivalité autour du test entre les Etats-Unis et la France, enfin l'utilisation des poches de sang contaminées. Je pense qu'on s'est à l'époque trompé sur la notion de séropositivité, Michèle Barzach vous le confirmerait.
Mme Emmanuelle Prada-Bordenave . Nous ne pouvons pas ici retracer toute l'affaire du sang contaminé. Je passe la parole au professeur Jean-Michel Dubernard.
M. Jean-Michel Dubernard, membre du collège de la Haute autorité de santé . Il faut en effet bien évaluer tous les aspects du principe de précaution.
Est-il logique que la thérapie cellulaire ou tissulaire soit assimilée au médicament ? Ne devrait-elle pas s'exercer dans un cadre spécifique ? Qu'en pensent les autorités de régulation ?
M. Jean Marimbert . Il ne vous a pas échappé que la France, dès 1996-1997, a développé un régime spécifique pour la thérapie cellulaire qui a été appliqué par l'Agence du médicament, puis par l'AFSSAPS qui lui a succédé, et qui n'était pas exactement calqué sur le régime du médicament. Notre pays s'était donc plutôt engagé dans la voie que vous souhaitez. Puis la régulation de la thérapie cellulaire s'est européanisée. Le règlement européen sur les médicaments de thérapie innovante du 13 novembre 2007 prévoit une procédure d'autorisation unique dans l'ensemble de l'Union. Il s'appliquera dans le futur à toutes les thérapies cellulaires, sauf celles conçues pour un malade déterminé, y compris industriellement dans les hôpitaux - il y aura là des exemptions. Le standard pour un essai multicentrique européen en thérapie cellulaire sera dorénavant d'être GMP ( Good Manufacturing Practice ). Ce n'est pas la France qui est à l'initiative de cette évolution qui crée une vraie difficulté, c'est l'Union européenne.
M. Jean-Michel Dubernard . Ne pourrait-on pas se battre à ce niveau ?
M. Jean Marimbert . Si. C'est un véritable enjeu. Le paradigme qui a prévalu pendant dix ans en France pour la thérapie cellulaire, avec ses succès, ses échecs, ses coups de frein, ses accélérations, n'était pas celui de la mise au point d'un médicament.
Mme Marina Cavazzana-Calvo . Je peux témoigner que l'Agence du médicament, puis l'AFSSAPS, loin de dresser des obstacles sur notre voie, ont accompagné les débuts de la thérapie cellulaire et génique en France, ce qui a permis des avancées thérapeutiques majeures. Hélas, aujourd'hui, au niveau européen, on veut imposer les mêmes normes que pour le médicament, ce qui est désastreux, surtout pour les recherches précliniques. Songez qu'on exige des conditions GMP pour la souris !
M. Olivier Boisteau . Il est impossible d'appliquer les GMP en thérapie génique autologue. En revanche, en allogénique, on va pouvoir se rapprocher, en tout cas s'inspirer de ce qui est fait pour la production d'anticorps avec la constitution de biobanques, l'amplification puis la purification des cellules. La solution serait de définir des GMP spécifiques pour la thérapie cellulaire. Mais ce ne serait pas une bonne idée que de s'en dispenser.
Les industriels du médicament ne cherchent pas à faire du « business ». Ils sont attachés au respect des valeurs éthiques. S'ils ne soutiennent pas davantage les recherches sur les cellules souches, c'est qu'ils ne sont pas encore en mesure de bien en évaluer l'intérêt thérapeutique. En revanche, ils sont intéressés par les avancées en thérapie cellulaire et par les cellules iPS qui constituent un outil extraordinaire pour tester la toxicité des substances pharmacologiques. Il ne s'agit pas de faire du « fric » - d'ailleurs pour les iPS, les brevets sont d'ores et déjà déposés - mais de mieux évaluer encore la toxicité des médicaments.
M. le président Claude Birraux . J'espère qu'il s'agit bien d'évaluer de nouveaux médicaments, et non d'offrir une nouvelle vie à des produits déjà anciens, sous un nouvel emballage !
Au terme de première cette table ronde, je remercie tous les participants.
DEUXIÈME TABLE
RONDE : LES ACTES MÉDICAUX
PRÉSIDENCE DE M. JEAN
MARIMBERT,
DIRECTEUR GÉNÉRAL DE L'AGENCE FRANÇAISE DE
SÉCURITÉ SANITAIRE DES PRODUITS DE SANTÉ
(AFSSAPS)
M. Jean Marimbert, directeur général de l'AFSSAPS. Mon mandat de directeur général de l'AFSSAPS touche à sa fin. Après ces sept années, je souhaite exposer en guise d'introduction certaines choses qui me tiennent à coeur.
Je suis persuadé qu'une régulation sanitaire peut faire une place à l'innovation, y compris l'innovation de rupture, dans le respect de sa mission de sécurité. Les exemples des « bébés bulles » et de la thérapie cellulaire de l'insuffisance cardiaque, présentés ce matin, ou celui du coeur artificiel implantable, que nous aborderons cet après-midi, l'illustrent bien.
C'est d'abord une affaire de volonté. J'en veux pour preuve le dispositif que nous avons mis en place pour être au contact des porteurs de projets innovants.
Ensuite, l'évaluation de la balance bénéfice/risque laisse des marges de manoeuvre. Autant l'analyse des données d'efficacité et de risque répond à des critères rigides, autant la balance elle-même - surtout pour le non-scientifique - est moins codifiée que l'on ne pourrait le croire. Son évaluation laisse une large place au jugement des experts. Elle peut même paraître un peu - un peu trop ? - impressionniste. Toutefois, elle permet d'accueillir davantage d'innovations véritables.
Par ailleurs, je souscris à la définition que Laurent Degos donne des sauts technologiques : bénéfice potentiel important pour la personne - qui, en l'occurrence, souhaite que l'on prenne des risques - et pour la collectivité, mais aussi degré de risque pas toujours évaluable et incertitudes quant aux risques qui pourraient surgir.
Pour autant, si la régulation sanitaire pose en effet des questions sociétales et implique un regard sociétal, ces situations ne doivent pas être placées hors de son champ. J'estime que l'on doit articuler et mettre en synergie une vision scientifique du bénéfice/risque et une vision sociétale apportée par des personnes autres que des scientifiques - notamment des éthiciens, mais pas seulement.
J'entends souvent que seuls les médecins peuvent exercer une responsabilité en matière de santé publique. Je doute que cet argument soit très convaincant. En effet, les non-médecins appelés à diriger de telles structures apportent parfois un regard plus permissif que celui des experts.
Il y a trois ans, après l'affaire d'Épinal, lorsqu'il s'est agi d'autoriser l'utilisation de cellules souches mésenchymateuses et alors que le rationnel scientifique était peu transposable à des personnes qui pourtant souffraient atrocement, les experts n'étaient pas enthousiastes. C'est le non-scientifique qui a pris la décision, en mettant en regard les souffrances endurées, les risques pas tout à fait maîtrisés, et enfin l'impuissance thérapeutique. Les résultats n'ont pas été formidables, mais on a au moins soulagé la douleur d'un patient. Cet exemple ne doit pas être pris pour de l'autoglorification. Il s'agit seulement de montrer que le regard d'un non-scientifique dans un processus de décision peut parfois ouvrir le jeu, et non pas le fermer.
La chirurgie comportementale, dont M. Luc Mallet va maintenant nous parler, s'est développée dans un cadre assez libre au départ et elle a donné des résultats prometteurs pour au moins un type d'affection. On se pose maintenant des questions quant à l'élargissement de son utilisation thérapeutique.
M. Luc Mallet, psychiatre, Centre de recherche de l'Institut du cerveau et de la moëlle épinière. Les techniques de stimulation cérébrale - ou, en d'autres termes, de neuromodulation implantée - permettent des progrès thérapeutiques considérables. Elles constituent également une révolution dans les neurosciences en ce qu'elles montrent que, contrairement au modèle hiérarchisé de MacLean où le cortex commande aux structures sous-corticales, les systèmes anatomiques qui gèrent la motricité, la pensée, les affects, sont largement partagés entre les zones.
Je passerai rapidement sur l'histoire de la recherche. Les techniques de lobotomie frontale ont valu à Egas Moniz le prix Nobel de médecine en 1949. Elles ont donné lieu à des excès aux États-Unis, où le psychiatre Walter Freeman opéra des milliers de patients de façon très contestable. L'arrivée de la chlorpromazine dans les années 1950 mit fin à la dérive de cette « psychochirurgie », tandis que se développait la chirurgie stéréotaxique, plus précise, et qui permettait de limiter les dégâts dans le cerveau.
L'histoire de la stimulation électrique du cerveau est quelque peu différente. Elle a commencé à la fin du XIX e siècle avec Hitzig et Fritsch. Le neurochirurgien canadien Wilder Penfield est à l'origine de progrès considérables. En France, à la fin des années 1980, Alim Louis Benabid a développé la technique de stimulation cérébrale profonde, qui constitue un saut conceptuel : on implante par voie stéréotaxique un « système embarqué » dans les structures profondes du cerveau. L'opération est l'occasion de réaliser des enregistrements électrophysiologiques servant au ciblage et à la caractérisation de certaines zones. L'électrode implantée est reliée par un câble sous-cutané à un générateur pilotable par télémétrie. On peut ainsi modifier les caractéristiques du courant délivré et moduler certains circuits de façon très sélective.
L'indication autour de laquelle cette technique s'est développée est la maladie de Parkinson. Aujourd'hui, plus de 80 000 patients dans le monde ont bénéficié d'implantations pour des indications neurologiques telles que tremblements essentiels, dystonie, maladie de Parkinson.
La recherche a pu ainsi observer les modifications comportementales intervenant lors de stimulations tests au bloc opératoire ou lors de modulations du site de stimulation. On a montré que le noyau sous-thalamique, que l'on considérait seulement comme une structure motrice, était en réalité impliqué dans la régulation du comportement et de certaines émotions : le système des ganglions de la base est un système majeur dans la régulation de certains aspects du comportement et de la cognition. Chez le primate, la perturbation réversible de l'activité des neurones dans ces mêmes zones, notamment les zones limbiques du globus pallidus externe, fait apparaître des comportements répétés. Même si les compulsions de lavage chez l'homme ne sont pas assimilables à de tels schémas, il s'agit cependant d'un début de modèle expérimental et pharmaco-anatomique, puisque la recherche sur le primate permet de pratiquer des thérapeutiques expérimentales.
Après que nous avons établi l'intérêt de moduler les activités des zones limbiques profondes, nous avons développé des protocoles concernant des maladies très handicapantes comme la maladie de Gilles de la Tourette. Les résultats sur la première patiente opérée, pour lesquels nous disposons de huit ans de recul, sont spectaculaires.
Pour ce qui est des troubles obsessionnels du comportement - TOC - sévères, comme les TOC de vérification, nous suivons des pistes de recherche anatomique impliquant certaines zones corticales, donc leurs corrélats sous-corticaux. Des travaux menés aux États-Unis visent à cibler les relations entre ces deux zones sur la base de constatations réalisées du temps de la psychochirurgie lésionnelle. En France, la recherche sur la stimulation dans les troubles du mouvement a donné lieu à des raisonnements plus physiopathologiques. Après avoir constaté la disparition de TOC chez des patients opérés pour une maladie de Parkinson, nous avons construit un protocole multi-centrique - le Comité consultatif national d'éthique, saisi par M. Benabid, avait alors validé la démarche. Lors de ces premières phases, nous avons suivi avec succès un protocole rigoureux en « double aveugle » montrant l'intérêt de la stimulation et nous n'avons rencontré aucun obstacle d'ordre réglementaire.
Un des intérêts majeurs de cette recherche est qu'elle modifie en amont la sélection des patients et la qualité des soins qu'on peut leur prodiguer. Bien qu'ils soient réversibles, les effets comportementaux indésirables révèlent l'étroitesse de la fenêtre thérapeutique dont nous disposons.
Ces recherches sont également importantes en ce qu'elles permettent d'effectuer des mesures électrophysiologiques lors d'expériences confrontant les patients à des situations expérimentales qui reproduisent les symptômes, et de mettre en évidence les activités neuronales liées à l'occurrence de ces symptômes. On pense même avoir mis en évidence pour la première fois des biomarqueurs électrophysiologiques corrélés à la sévérité de l'affection et à la réponse à la stimulation. Certaines structures des ganglions de la base semblent bien être au coeur du processus obsessionnel qui accompagne ces maladies.
Au total, la recherche a rencontré un succès important pour une cible déterminée. D'autres travaux se révèlent intéressants pour d'autres cibles. En France, trois protocoles sont en cours concernant les TOC. Le premier tend à évaluer l'intérêt de la stimulation d'un seul côté du cerveau, ce qui permettrait de diminuer le risque clinique. Le deuxième, qui a bénéficié du programme de soutien aux techniques innovantes coûteuses, vise à comparer la cible sous-thalamique à la cible accumbens dans un objectif médico-économique de généralisation d'une technique. Le troisième, qui consiste à implanter chez le même patient des électrodes visant ces deux cibles, est la seule façon de poser certaines questions physiopathologiques.
Il a été très difficile de déposer ce dernier protocole. Nous n'avions aucun soutien industriel. Après trois ans de travail, nous nous sommes heurtés à l'AFSSAPS, qui nous a opposé son évaluation du rapport bénéfice/risque. J'ai contesté cet avis ligne par ligne et ai fini par obtenir gain de cause. La recherche est à un moment important, avec la perspective d'un élargissement des indications à la dépression et aux addictions, mais nous touchons aux limites de ce qui nous permettrait d'aller plus loin.
M. Jean Marimbert. Le sujet que va nous présenter maintenant Mme Mandelbaum, la vitrification ovocytaire, semble moins marqué par l'ampleur des difficultés scientifiques que par des difficultés purement juridiques.
Mme Jacqueline Mandelbaum, biologiste de la reproduction, chef du service d'histologie, biologie de la reproduction / CECOS (Centre d'étude et de conservation des oeufs et du sperme humains) de l'hôpital Tenon, membre du Conseil d'orientation de l'Agence de biomédecine. J'élargirai quelque peu le thème proposé et parlerai des sauts technologiques en AMP - assistance médicale à la procréation.
La fécondation in vitro représente en elle-même un saut technologique résultant d'une démarche d'expérimentation animale ancienne. En 1880, à Vienne, Schenk réalise les premiers essais de culture in vitro de l'embryon de mammifère conçu in vivo. En 1944, Rock et Menkin mènent les premiers essais d'insémination in vitro d'ovocytes humains, mais c'est un échec : les trois embryons qu'ils pensent avoir obtenus sont des ovocytes fragmentés. C'est en 1954 qu'ont lieu les premières fécondations in vitro authentifiées, pratiquées sur des ovocytes de lapines, par l'équipe française de Charles Thibaut à Jouy-en-Josas et par l'équipe américaine de Chang. Les premières naissances interviennent en 1959.

Présentation de Mme Jacqueline Mandelbaum
En 1965, Robert Geoffrey Edwards s'attelle à la maturation in vitro en réalisant une étude comparative chez la souris, la brebis, la vache, le porc, le singe rhésus et l'humain. Il lui faudra six ans de recherche pour obtenir la fécondation et le développement de l'embryon humain in vitro. Il décrit alors tous les stades de clivage de l'embryon humain, jusqu'au stade de blastocyste expansé puis éclos. Edwards étudie la normalité de ces embryons non seulement d'un point de vue morphologique et cinétique, mais aussi de leur équipement cytogénétique.
Il lui faut encore sept ans d'essais cliniques, dans un environnement scientifique, médical et sociétal très hostile, pour arriver enfin à la naissance de Louise Brown le 25 juillet 1978. Elle est le soixante-quinzième être vivant conçu par cette technique dans le monde.

Présentation de Mme Jacqueline Mandelbaum
Il s'agit incontestablement d'un saut technologique pour l'espèce humaine, précédé par une large expérimentation animale. C'est aussi un progrès thérapeutique majeur puisque entre 1 et 3 % des enfants naissent dans le monde grâce à ces méthodes et qu'ils représentent à ce jour environ 4 millions d'êtres humains. Trente-deux ans après, en 2010, le prix Nobel de physiologie et de médecine vient récompenser ces recherches. Comme le dit Martin Johnson, un élève de Bob Edwards, mieux vaut tard que jamais !
Quelques mois avant la naissance de Louise Brown, j'avais présenté un dossier de candidature à l'INSERM auprès d'Étienne Beaulieu. Celui-ci avait salué mon travail sur la fécondation in vitro des ovocytes de hamsters, mais il s'était opposé à des recherches sur l'ovocyte humain, insistant sur la nécessité de développer un modèle sur les primates.
La naissance de Petri, singe rhésus issu d'une fécondation in vitro, survint six ans après celle de Louise Brown, alors que les premiers travaux de Bob Edwards sur la maturation ovocytaire chez le singe rhésus datent de 1965 et que, durant la même période, huit groupes de recherche travaillaient dans le monde sur la thématique de la fécondation in vitro chez le primate. Si Bob Edwards avait suivi les conseils du Medical Research Council et avait continué de travailler sur le primate, il n'est pas certain qu'il aurait obtenu le même succès car il tirait sa motivation d'un moteur très puissant : le désir de traiter la stérilité tubaire, et les motivations associées des couples. Martin Johnson a publié dans Human Reproduction un article éclairant sur la manière dont la recherche institutionnelle a refusé de subventionner ces travaux, lesquels ont été entièrement menés sur des fonds privés.
Deuxième exemple de saut technologique : la congélation embryonnaire.
Cette méthode de conservation est essentiellement obtenue par une congélation lente où l'effet délétère de la cristallisation est limité par l'ajout de cryoprotecteurs et par un contrôle des températures de refroidissement et de réchauffement. Elle s'appuie là aussi sur une large expérimentation animale. Les premières congélations de morulae de souris sont réalisées par Whittingham en 1972. Comme pour la fécondation in vitro, après des essais précliniques chez l'homme, les premières naissances sont obtenues en 2004 en Australie et aux Pays-Bas. Depuis, cette technique permet de conserver les embryons surnuméraires - 25 à 50 % des tentatives - et de limiter le nombre d'embryons transférés ainsi que le risque de grossesse multiple. Elle est un atout indispensable pour développer le transfert d'embryon unique - le fait de disposer d'embryons congelés permet 12 % de naissances additionnelles par ponction. Elle peut être appliquée à tous les stades de développement embryonnaire, du zygote au blastocyste. Il s'agit d'un progrès thérapeutique majeur. En 2008, 15 % des enfants nés après FIV ICSI - fécondation in vitro par injection intracytoplasmique - en France avait fait l'objet d'une congélation embryonnaire.
Troisième exemple : la micro-injection intracytoplasmique d'un spermatozoïde, ou ICSI.
Contrairement à une idée reçue, cette technique a fait l'objet d'expérimentations préalables chez l'animal. En 1983, Markert a présenté un travail qui consistait à injecter des têtes de spermatozoïdes dans des ovocytes de souris. La difficulté rencontrée est que la majorité des ovocytes étaient dégénératifs après cette effraction du cytoplasme. Sur les ovocytes restants, on n'a pu obtenir que moins de 3 % de naissances.
En 1990, des expériences sur des taureaux dont la qualité spermatique était mauvaise ont montré que cette procédure améliorait le taux de fécondation.
En 1991, on améliore le taux de clivage chez le lapin à partir de spermatozoïdes testiculaires, mais sans obtenir aucune grossesse.
En 1992, d'autres expériences menées sur le hamster se traduisent par des résultats très mitigés pour ce qui est du taux de fécondation et du taux d'ovocytes dégénératifs.
On a également mené des essais précliniques chez l'humain. Les travaux de Lanzendorf en 1988 et de Ng en 1991 font apparaître également un fort taux d'ovocytes dégénératifs et des taux de fécondation très moyens. Devant ces résultats décevants, on s'oriente vers l'injection de spermatozoïdes dans l'espace périvitellin, entre la zone pellucide et le cytoplasme - technique dite SUZI, ou subzonal insemination.
C'est alors qu'intervient l'erreur féconde de Gianpiero Palermo, un chercheur italien travaillant dans l'équipe d'André Van Steirteghem à Bruxelles : en utilisant une pipette d'injection plus acérée, il lui arrivait parfois de traverser involontairement la membrane plasmique et de réaliser une injection intracytoplasmique. Il constata que les ovocytes étaient fécondés avec une bien plus grande fréquence de cette manière que par SUZI, et que le développement embryonnaire qui s'ensuivait était normal. Sur la base de ces résultats, il décide de partager les cohortes ovocytaires pour 11 cycles : une moitié en ICSI, une moitié en SUZI. Le résultat ne laisse aucun doute : le taux de fécondation est de 4 % en SUZI, de près de 40 % en ICSI. L'expérience de l'ICSI se poursuit et une publication de The Lancet de juillet 1992 décrit les trois premières naissances de quatre enfants.
L'équipe mène ensuite 300 cycles comparatifs qui montrent toujours un bien meilleur taux de fécondation en ICSI - 59 % contre 18 % en SUZI - et des taux de grossesse cinq fois plus importants. Le SUZI est alors abandonné définitivement au profit de l'ICSI.
L'ICSI représente un saut technique majeur, immédiatement reproductible et diffusible, reconnu par la communauté scientifique. Les équipes viennent à Bruxelles, observent ce qui se fait, le transposent dans leurs laboratoires et obtiennent les mêmes résultats.
Il apparaît clairement que c'est pour l'espèce humaine que l'ICSI a été le plus simple. Il faut attendre 1995 et la mise en oeuvre de techniques particulières d'injection piézoélectrique pour obtenir la même efficacité chez la souris, dont le spermatozoïde a un flagelle très long, et où la destruction cytoplasmique était très importante avant que l'on ne trouve des astuces technologiques. En d'autres termes, la réalisation de l'ICSI chez l'humain a permis de faire progresser l'ICSI pour d'autres espèces. Bref, s'il est bien entendu très important de disposer de modèles, ceux-ci ne sont pas toujours transposables.
L'ICSI est un progrès thérapeutique incontestable non seulement sur le plan individuel mais aussi sur le plan de la santé publique : cette technique a constitué une révolution dans le traitement des stérilités masculines, qui représentent 50 % des stérilités du couple et 60 % des ponctions en AMP.
Quant au risque, l'étude menée sur les enfants nés après ICSI ne montre pas de différence notable par rapport aux enfants issus d'une fécondation in vitro. L'équipe de Bruxelles dispose des données relatives au suivi d'enfants dont l'âge va jusqu'à 14 ans. Pour les garçons, en particulier, la puberté se déroule de façon identique à celle d'une population née par grossesse spontanée.
La fécondation in vitro humaine, la congélation embryonnaire ou l'ICSI auraient-elles pu être autorisées et financées dans le cadre actuel de la recherche en AMP ? Pour illustrer cette question, j'en viens à l'exemple de la vitrification ovocytaire.
La vitrification est une technique différente de congélation où, grâce à des concentrations accrues de cryoprotecteurs et des vitesses de refroidissement très élevées, on passe directement d'une phase liquide à une phase amorphe, vitreuse, sans modification de l'organisation moléculaire et sans cristal. C'est une technique ancienne, décrite en 1985 par Rall et Fahy pour l'embryon de la souris, et en 1989 pour l'ovocyte. Des naissances normales ont été obtenues chez la souris, le lapin, le rat, le mouton, le bovin - technique développée par Massip en Belgique -, et l'humain. En 2009, l'étude de la chercheuse suédoise Wennerholm ne révèle aucune différence avec une technique de congélation lente pour ce qui est de la qualité des enfants nés et du taux de malformation.
Si la vitrification a été peu utilisée chez l'humain jusqu'en 2001, c'est qu'elle n'était pas plus efficace, voire moins, sauf lorsque l'on ménageait un contact direct avec l'azote liquide. Mais la conservation devenait difficile et posait des problèmes de sécurité sanitaire évidents.
Cependant, le développement de nouveaux supports a permis de relancer cette méthode qui a montré sa supériorité pour la congélation du blastocyste et d'ovocytes matures de l'espèce humaine. La congélation lente, pratiquée depuis 1987, présentait un faible taux de succès mais a permis la naissance de plusieurs centaines d'enfants. L'étude d'une chercheuse espagnole, Cobo, menée sur 600 receveuses montre qu'il n'existe aucune différence entre les ovocytes frais et les ovocytes vitrifiés. Rienzi, en 2010, obtient les mêmes résultats en fécondation in vitro intraconjugale. L'étude de Chian sur 200 enfants nés après vitrification ovocytaire montre qu'il n'y a pas de surcroît d'anomalies. Nous ne disposons pas encore d'étude comparative avec une population témoin mais ces résultats sont très rassurants.
Dans ce domaine, il existe une exception française. En juillet 2008, un protocole de recherche biomédicale est déposé pour étudier la faisabilité de la vitrification ovocytaire dans la pratique de l'AMP. Deux ans plus tard, après étude par l'AFSSAPS, le ministère oppose un refus et interdit la vitrification, y compris pour d'autres équipes. Sans doute pourrez-vous expliquer mieux que moi les tenants et les aboutissants de cette décision, monsieur Marimbert. Ce que l'on m'en a dit, c'est que, dès lors qu'il y avait demande d'autorisation, on a considéré que c'était une technique innovante, non validée, qui allait aboutir à la création d'embryons. Comme les embryons qui ont fait l'objet d'une recherche ne peuvent pas être transférés, l'interdiction a été prononcée.
Dans ces conditions, la fécondation in vitro, la congélation embryonnaire ou l'ICSI n'auraient sans doute pas été autorisées.
La conséquence, c'est que les équipes se replient sur des thématiques moins problématiques ou plus fondamentales, laissant aux autres pays le champ de l'innovation technologique que nous utiliserons ensuite « clés en main ».
La vitrification ovocytaire, qu'il n'y a aucune raison de ne pas autoriser au vu de l'expérience acquise dans d'autres pays chez l'animal et chez l'humain, n'est pas à proprement parler un saut technologique. Cependant, il s'agit d'une amélioration technique qui rendra ponctuellement des services dans la préservation de la fertilité, la gestion du don d'ovocytes, la stratégie de prise en charge des couples en AMP, ce qui est déjà considérable !
J'entends beaucoup dire que l'on va utiliser cette interdiction pour diminuer, voire interdire, la congélation embryonnaire, ce qui serait une véritable régression thérapeutique. L'Italie, qui a déjà connu ce parcours, vient d'autoriser de nouveau une congélation embryonnaire a minima. Les centres les plus en pointe, comme celui de Laura Rienzi, n'utilisent la congélation d'une partie des ovocytes que pour 40 % des ponctions. Il est préférable de laisser vivre toutes les possibilités afin que chacun puisse les utiliser.
Une politique dynamique d'innovation passe par le renforcement de la recherche animale en reproduction et l'interaction de cette recherche avec les équipes d'AMP. La méthode la plus utilisée chez l'humain, la congélation lente, a été mise au point avec des chercheurs de l'INRA à Jouy-en-Josas. En revanche, la technique de vitrification a été développée ailleurs, sans doute parce que les équipes zoologiques avaient d'autres terrains d'action et insuffisamment de personnes pour s'intéresser à la question.
Il conviendrait également d'améliorer la lisibilité de la réglementation de la recherche en AMP en clarifiant les exigences en matière de nouvelles techniques et en fluidifiant le parcours entre les différentes agences. L'AMP est une thérapeutique. L'interdiction de toute innovation technologique dès lors qu'elle aboutit à la constitution d'embryons équivaut à une condamnation de la discipline. Avec l'accord des couples, l'embryon devrait pouvoir bénéficier de ces thérapeutiques comme l'adulte qu'il deviendra peut-être après transfert. Une recherche sur l'embryon autorisée mais encadrée lèverait le discrédit qui entoure aujourd'hui le sujet, et permettrait de réfléchir plus sereinement à un parcours plus favorable à l'innovation, fondé sur l'expérimentation animale et comprenant des essais précliniques avec l'accord des couples concernés. On peut penser à une procédure d'attente pour des embryons ayant subi un check-up de non-anomalie - comme cela se pratique pour le diagnostic préimplantatoire - puis à des essais cliniques.
M. Jean Marimbert. La notion d'« erreur féconde », que vous avez mentionnée, est insuffisamment en usage en France, me semble-t-il.
Par ailleurs, je confirme que le blocage autour de la vitrification ovocytaire depuis deux ou trois ans n'est pas dû à la régulation exercée par les agences. En effet, L'AFSSAPS et l'ABM étaient tout à fait favorables à une interprétation souple de la loi et à l'évaluation des dossiers sur leurs mérites scientifiques. Il s'agit bien d'un blocage juridique qu'il appartient au Parlement de lever s'il le souhaite.
M. Elias Zerhouni, professeur de radiologie, président Monde, recherche et développement, en charge des médicaments et des vaccins, Sanofi-Aventis. Que ce soit en termes de santé, de recherche biomédicale ou du point de vue sociétal, nous vivons une période extraordinaire caractérisée par des tensions et des antagonismes.
Partout dans le monde, les États ont de plus en plus de mal à faire face à l'augmentation des coûts de santé, que les hommes politiques et les économistes attribuent un peu vite à l'innovation technologique. En fait, à cause de changements démographiques et épidémiologiques, les maladies aiguës, à court terme, létales - principalement cardiovasculaires, comme l'infarctus du myocarde ou l'ictus cérébral -, qui dominaient il y a quarante ans et en fonction desquelles le système de santé avait été mis en place, ont diminué au profit des maladies chroniques, qui représentent désormais 75 % des coûts de santé dans presque tous les pays développés et apparaissent dans les pays en voie de développement. Les innovateurs doivent se focaliser sur ces facteurs, d'autant que 80 % desdits coûts concernent 20 % des malades. Si l'on ne tient pas compte de cette situation, certaines pressions sociales et budgétaires risquent de s'aggraver et de peser sur la capacité des pays à allouer des ressources aux activités innovantes.
Des tensions proviennent également du vieillissement de la population et du développement de nouvelles pathologies. L'OMS prévoit par exemple que l'obésité, facteur de dépression, sera bientôt la première cause d'incapacité de travail dans le monde entier pour la tranche d'âge comprise entre 25 et 44 ans. Ces évolutions démographiques appelleront nécessairement de nouvelles politiques et des stratégies innovantes. Il n'y a pas d'autre choix que de réunir toutes les forces sociales, scientifiques et technologiques afin de coordonner les efforts pour trouver des solutions. Le besoin d'innovation, qui peut s'analyser comme un facteur négatif en termes de coûts mais positif sur le plan des soins, impose d'adopter de nouvelles stratégies non seulement biologiques mais aussi technologiques et sociales. Il faut redéfinir de manière générale le rapport entre, d'une part, la valeur médicale relative d'une innovation et, d'autre part, ses risques, ses bénéfices et ses coûts. À l'échelle du monde, on doit travailler non plus sur le rapport entre risque et résultat ( risk/benefit) ou sécurité et efficacité ( safety/efficacy ), mais sur un nouvel index de valeurs médicales encore à créer, qui supposera une entente à tous les échelons et intégrera la participation du public. Je rejoins M. Marimbert : il faut éviter le débat d'experts et ouvrir un débat social.
Du point de vue de l'innovation elle-même, il faut aussi changer de paradigme. Actuellement, on intervient très tard dans le développement de la maladie et pour un coût très élevé. Puisque les maladies chroniques, qui pèsent de plus en plus sur le secteur médical, débutent physiologiquement vingt ans avant leur déclenchement clinique, la recherche doit cesser de se concentrer sur un seul stade de la maladie pour s'étendre à l'ensemble du cycle biologique. Passer du curatif au préemptif implique qu'à l'échelle biologique, on essaiera de comprendre la complexité des maladies et leur évolution, afin d'intervenir de plus en plus tôt. Ce faisant, on ruinera l'idée reçue selon laquelle plus la médecine réussit, plus elle coûte cher et plus elle sera inaccessible. Réfutant ce paradoxe qui, à terme, étoufferait l'innovation, le débat doit encourager une stratégie sociopolitique tournée vers la compréhension de la complexité biologique. Il faut investir dans ce domaine en sciences fondamentales.
Après avoir dirigé le National Institute of Health (Institut national de la santé), qui couvre l'ensemble de la recherche biomédicale aux États-Unis et gère un budget de 30 milliards de dollars, j'ai été frappé du fait que certaines avancées scientifiques extraordinaires comme le séquençage du génome humain ou le développement des méthodes transgéniques ne se traduisent pas par l'arrivée sur le terrain des nouvelles thérapies pourtant très efficaces. Cette année, le Food and Drug Administration (FDA) a approuvé 24 nouvelles molécules - dont 7 ou 8 vraiment originales -, contre 50 en 1996. C'est une autre tension ou un autre paradoxe : l'augmentation des investissements en recherche et développement aboutit à une moindre productivité, ce qui s'explique non par un défaut d'organisation ou de créativité des entreprises académiques ou privées, mais par un manque fondamental de connaissances permettant d'éclairer la complexité biologique.
L'article sur la stimulation cérébrale paru dans le New England Journal Of Medicine, qu'a présenté M. Mallet, compte une trentaine d'auteurs, ce qui prouve que la complexité des problèmes renvoie à celle des équipes. On ne peut plus faire de progrès en médecine appliquée sans une stratégie destinée à encourager l'innovation multidisciplinaire à l'échelle appropriée. Tous nos systèmes de régulation et de supervision sociales doivent s'adapter à cette réalité. Si, dans les prochaines décennies, nous ne réglons pas le problème de la complexité biologique, nous serons de moins en moins capables d'intervenir avec un index médical de valeurs suffisamment justifié.
Il faut donc envisager une nouvelle médecine, qui reposerait sur quatre nouveaux piliers. Elle doit d'abord être prédictive et individualisée, c'est-à-dire capable, dans le domaine biologique, de prédire le développement potentiel de la maladie chez un individu. Même si nous progressons à cet égard - en cancérologie, les signatures génétiques et protéomiques de certaines tumeurs dictent des thérapies personnalisées différentes -, nous sommes encore loin du but. Ainsi, en santé publique, on traite beaucoup de malades qui ont un taux de cholestérol élevé avec des statines, qui étaient jadis très onéreuses. Or, sur 100 malades traités, seuls 8 ou 10 ont réellement besoin de tels médicaments, les autres n'étant pas exposés au risque d'artériosclérose. L'incapacité à disposer de biomarqueurs qui stratifient cette population nous contraint à dépenser dix fois trop.
À la prédictivité et à la personnalisation, s'ajoute un troisième pilier, qui constitue un grand espoir : une compréhension de la complexité qui permette de préempter la maladie, comme le fait le vaccin contre le cancer du col. Ces trois piliers amèneront la médecine à traiter la maladie à un stade plus précoce où les patients ne sont pas véritablement des malades.
Ce changement fondamental de notre conception du système de santé met en jeu un dernier pilier : une plus grande participation des personnes aux soins et une redistribution des ressources. Celles-ci seront guidées par des avancées technologiques au terme desquelles les interventions médicales voire chirurgicales seront de moins en moins invasives. Les technologies de communication et le biosensing visant à mesurer les paramètres physiologiques permettront de traiter les malades à domicile, surtout s'ils souffrent de maladies chroniques. La possibilité de mesurer les paramètres essentiels, comme l'hypertension et le taux de glucose, débouchera sur des traitements dont la conformité sera plus effective ou, en d'autres termes, qui seront mieux suivis. Ces avancées technologiques font entrevoir un monde où le nombre de centres médicaux chers et compliqués diminuera au profit de centres mieux distribués, où l'on pratiquera une médecine participative pour un coût moindre et un bénéfice réel supérieur.
DÉBAT
M. Jean Marimbert. Vous avez souligné que l'évolution de l'épidémiologie induit un changement de paradigme tant dans le développement que dans la réception de l'innovation, ce qui ne sera pas sans incidences sur la régulation et sur l'allocation des ressources, dans un contexte où elles restent rares.
Le président Claude Birraux. M. Zerhouni a posé des questions d'une grande actualité. Selon M. Claude Huriet, président de l'Institut Curie, les recherches qu'il vient d'évoquer sont freinées par certains articles de loi et par les décisions de la CNIL. Le problème a été soulevé récemment lors d'un congrès organisé par M. Alain Huriet sur la médecine personnalisée.
M. Jean de Kervasdoué. On surévalue généralement l'impact du progrès technique sur les dépenses de santé, qui se limite en moyenne à 0,2 %. Au Japon, pays du monde où l'on vit le plus vieux, elles représentent 8,5 % du PIB, alors même que les Japonais recourent six fois plus que les Français aux scanners et aux IRM, et consomment beaucoup de médicaments. L'augmentation des dépenses de santé tient surtout à l'importance des maladies chroniques, qui représentent 50 % des dépenses dans les pays du Maghreb et plus de 70 % dans le nôtre, où le vieillissement n'influe que de 0,5 % sur l'augmentation du budget de la santé.
Aujourd'hui, la France, dont le système de santé est le deuxième au monde, dépense en moyenne 57 milliards d'euros de plus que la moyenne des pays européens. Il y a quarante ans, son budget pour la santé s'élevait à 5,7 % du PIB, contre 6,3 % en Suède, ce qui constituait le record mondial. Il est aujourd'hui de 11,7 % en France, contre 9 % en Suède, différence considérable puisqu'un point de PIB représente 20 milliards d'euros. Le dérapage est dû à des causes spécifiques qui ne doivent rien à la fatalité, contrairement à ce que prétendent tous les ministres, de droite comme de gauche.
M. Elias Zerhouni. Les statistiques générales sur les coûts de santé et la durée de la vie cachent des réalités nationales très différentes. Aux États-Unis, si l'on fait abstraction de la mortalité due aux armes à feu et aux naissances avant terme chez les jeunes filles non mariées, les statistiques sur la longévité sont les mêmes que dans les autres pays. Au Japon, les familles ne déclarent pas toujours le décès d'un proche au moment où il survient, pour continuer à percevoir certaines allocations, ce qui fausse les chiffres. Autant de facteurs culturels qu'il importe de prendre en compte.
Contrairement à ce que prétend Peter Orszag, directeur du Bureau de la gestion et du budget aux États-Unis, selon lequel 50 % des dépenses de santé seraient directement imputables à l'innovation, c'est en fait une mauvaise utilisation de celle-ci qui explique l'augmentation des coûts, lesquels seraient réduits par un diagnostic ciblé, personnalisé et précis.
M. le président Claude Birraux. On peut se demander si notre système de santé administré ne renchérit pas certains coûts. Je me souviens qu'un hôpital avait dû quasiment faire l'aumône parce que la DDASS ou la DRASS lui refusaient l'acquisition d'un scanner, matériel qu'on trouve aujourd'hui même dans les petits hôpitaux, parce que la multiplication des achats a fait baisser les prix.
Parallèlement à des centres de santé de plus en plus grands, on pourrait imaginer de petites unités irriguant le tissu par capillarité. Ainsi, une personne issue du CERN avait projeté d'installer dans un camion semblable à ceux qu'on utilise pour la transfusion sanguine un matériel permettant de réaliser des IRM, pour mener une campagne itinérante dans les petits hôpitaux.
M. Elias Zerhouni. Si les ressources ne sont pas toujours bien distribuées, c'est que l'implantation des systèmes de santé, très politisée, ne dépend pas toujours de critères opérationnels. Certaines distorsions s'expliquent par la demande des contribuables, qui attendent une présence et une réactivité ; d'autres, par le fait que beaucoup de décisions sont prises à l'échelle centrale par quelqu'un qui se comporte davantage en ministre des maladies qu'en ministre de la santé. C'est précisément ce paradigme qui doit changer.
M. Jean Marimbert. L'évolution de la prise en charge des pathologies - par exemple, le traitement en ville de nombre de maladies chroniques autrefois traitées à l'hôpital - est un nouveau défi pour les régulateurs. Traditionnellement, les signalements de pharmacovigilance, qui sont sous-notifiés, émanaient de l'hôpital. Ils représentent désormais un enjeu pour la médecine de ville.
M. Luc Mallet. Comme l'a souligné M. Zerhouni, l'étape suivante de nos recherches consiste à retourner vers le préclinique et l'animal, et à poser de nouvelles hypothèses originales de psychologie expérimentale. Il faut faire exploser les concepts à partir de ce que nous aurons observé en stimulation.
L'expert de l'AFSSAPS qui a rédigé l'avis motivé visant à m'interdire le protocole que je mène actuellement, contestait qu'on puisse proposer une double implantation - soit quatre électrodes au lieu de deux - chez les patients. Dans le modèle du TOC, il existe aujourd'hui deux pistes différentes, dont le coût est difficile à évaluer. Le premier protocole est mené de manière multicentrique, ce qui complique sa gestion. Le second, qui consiste à implanter deux électrodes chez le même patient, réclame une plus haute technicité mais porte sur beaucoup moins de patients. Il ne peut pas être mené partout et suppose des moyens multidisciplinaires beaucoup plus importants. Je considère cependant que les connaissances qu'il apportera permettront de le rentabiliser. Ce protocole peut apporter un gain commun très supérieur au risque personnel, lequel est évalué par ailleurs et fait l'objet de toutes les réglementations. Les promoteurs s'étaient déjà montrés très exigeants. En l'occurrence, il s'agit d'institutionnels, issus de l'AP-HP, car, dans le domaine du matériel implanté intracérébral, les industriels n'apportent aucune aide - même si, de manière assez mystérieuse, ils disposent ensuite de marquage CE -, et ne prennent aucun risque.
À l'issue de la discussion, l'AFSSAPS a reculé. Peut-être a-t-elle été convaincue par mes arguments explicites, puisque j'ai défendu pied à pied la technique et la méthodologie envisagée. Mais peut-être sont-ce mes arguments implicites qui ont prévalu : dans l'état actuel de l'expertise en France et compte tenu du nombre d'équipes impliquées, il me semble impossible de s'en remettre à un expert qui ne soit pas exposé à des conflits d'intérêts, ce qui supposerait qu'on délocalise l'expertise au niveau européen.
Comme l'a suggéré M. Zerhouni, les deux niveaux d'investigation ont été mal évalués. Les protocoles multicentriques n'ont rencontré aucun problème d'autorisation et offriront aux industriels la possibilité d'être remboursés. Nous ne nous plaçons pas dans ce cadre, mais nos travaux, dont les conséquences sont très importantes en termes de recherches et d'application, déboucheront peut-être sur d'autres techniques non invasives. En matière de connaissances sur la pathologie, les protocoles que nous proposons offriront un retour sur investissement considérable, même s'ils sortent de ce qu'on évalue habituellement en recherche clinique.
M. Bernard Avouac. Même s'il peut sembler difficile à un représentant des industriels de soutenir l'idée qu'il faut augmenter la prise de risque, je vais m'y employer. Alors que les autorités sont habituées à évaluer le rapport entre bénéfice et risque - sachant qu'il n'y a jamais assez de données pour asseoir le bénéfice, tandis que le risque est toujours possible, en d'autres termes que seul le risque est sûr -, l'innovation et les sauts technologiques font intervenir une balance originale entre un bénéfice potentiel et un risque imprévisible.
Depuis longtemps, nous pensons qu'il faut plaider pour un principe de précaution inverse. Quand un risque est maîtrisé, de quel droit interdirait-on au patient de bénéficier d'un moyen thérapeutique qui ne peut être que supérieur à ceux qui existent ? Dès lors que le risque est assumé, c'est le rapport perte de chance vs risque qu'il faut évaluer. Il s'agit non d'être laxiste, mais de réguler la balance entre le bénéfice et le risque de l'innovation. Nous devons imposer à cet égard une nouvelle vision, contraire à celle qui s'impose actuellement. De même, il faut combattre l'idée, séduisante en période de budget contraint, selon laquelle l'innovation coûterait cher, et qu'y renoncer permettrait de réaliser des économies.
M. René Frydman. Dans mon domaine, les chercheurs ont souffert d'un blocage législatif : l'interdiction de toute recherche sur l'embryon - sauf de manière dérogatoire et pour un temps limité - les a empêchés de faire évaluer scientifiquement leurs projets. Sans doute faut-il distinguer la recherche sur l'embryon hors de tout projet parental, laquelle aboutit habituellement à la destruction de l'embryon dans un cadre cerné, et les travaux menés dans le cadre d'une innovation thérapeutique, qui visent à soigner non l'embryon mais la stérilité du couple. Le débat que nous menons depuis trois ans avec les instances concernées a empêché toute recherche sur la vitrification. Même si cette technique n'est pas la panacée, il est regrettable qu'en France, on ne puisse pas avancer dans ce domaine.
Même s'ils ont souligné les points positifs d'une régulation qu'ils souhaitent depuis l'origine, et qui garantit transparence et évaluation, tous les intervenants déplorent l'importance des freins législatifs et administratifs qui s'ajoutent à la difficulté intrinsèque des sujets. Nous renvoyons ce problème législatif à la représentation nationale. Peut-elle donner une impulsion à l'innovation en agissant sur la régulation ? Les chercheurs ont l'impression d'avancer dans un maquis de procédures, qui, même quand elles résultent d'excellentes intentions, aboutit à un blocage. Tous ceux qui ont innové - le professeur Pouliquen sur la cataracte, le professeur Carpentier sur les implantations cardiaques ou moi-même sur la fécondation in vitro - reconnaissent que les recherches qu'ils ont entreprises ne seraient plus possibles aujourd'hui. Ainsi s'explique la déception des chercheurs et des médecins, dont certains ont commencé à s'expatrier. Pour lever ce frein sans tomber dans des dérives commerciales ou dans un laxisme que nul ne songe à défendre, il faut adopter une régulation équilibrée - qui suppose la transparence et l'évaluation a posteriori - et renoncer aux déclarations a priori, qui n'ont rien de scientifique, puisque le rôle de la science est précisément de tester et d'évaluer. Pourquoi limiter la recherche aux iPS en interdisant toute recherche sur les cellules souches embryonnaires ? Si cette interdiction relève d'une position philosophique, elle pose problème aux scientifiques car enfin, s'il était démontré que les iPS étaient valables sur le plan thérapeutique, nul ne s'acharnerait à vouloir sortir des cellules souches embryonnaires. L'évaluation est indispensable dans un souci d'objectivité.
Au problème législatif s'ajoute un problème administratif. Quand un chercheur a une idée, il lui faut plusieurs mois pour affiner son projet, après quoi il devra passer successivement devant un comité de protection des personnes (CPP), puis devant l'ABM, puis devant l'AFSSAPS. Il peut aussi apprendre in fine - c'est ce qui s'est passé dans le cas de la vitrification - qu'un problème juridique va bloquer son projet pendant deux ans. Après six mois d'élaboration et un an et demi de procédure, auxquels s'ajoute, outre un ou deux ans pour réaliser le projet, le temps d'écrire un article et de l'envoyer à des revues, il constatera que quelqu'un l'aura fait paraître avant lui, car les idées sont souvent partagées par la communauté scientifique.
S'il faut donner une impulsion, c'est aussi à ce niveau, en regroupant les différents organismes. Quand bien même leur appréciation en matière d'éthique, de toxicité, de faisabilité ou d'évaluation scientifique est légitime, ils devraient apporter une réponse immédiate et globale, seul moyen de raccourcir les délais. Le moment est venu de redresser la situation. À quoi bon mettre les chercheurs en vedette dans le cadre d'une fête de la science alors qu'on bloque chacune de leurs demandes ?
M. Elias Zerhouni. Je partage l'avis de M. Frydman. Cela dit, le problème est mondial. J'ai fait réaliser aux États-Unis une étude sur l'allégement régulatoire. Le fait que l'instrument législatif soit créé sans grand souci d'harmonisation pour répondre à des crises produit des effets délétères.
Dans les années 1970, un doctorat en sciences médicales réservé aux médecins et chercheurs doctorants a été lancé aux États-Unis. Son bilan est effrayant. En 1980, 20 % d'entre eux se consacraient à la recherche fondamentale dans les laboratoires, avec des modèles animaux et 80 % à la recherche translationnelle visant à régler un problème de biologie humaine. Aujourd'hui, 85 % se sont dirigés vers les sciences de base, où le problème de la régulation ne se pose pas. Il est en effet moins productif, en matière de carrière, de passer cinq ans sur un papier que d'en publier dix pendant la même période. Ces distorsions creusent un fossé entre les chercheurs et découragent les jeunes médecins. Sans remettre en cause le besoin d'éthique, il est essentiel de mener une réflexion ouverte qui prenne en compte la différence entre l'innovation créatrice, l'innovation de diffusion et l'innovation d'application.
Mme Jacqueline Mandelbaum. Comme M. Frydman, je plaide pour l'autorisation d'une recherche encadrée sur l'embryon. L'interdiction, qui jette l'opprobre sur les travaux qui pourraient être effectués dans notre discipline, constitue un frein à tous les protocoles que nous présentons. L'autorisation encadrée constituerait un cadre plus serein, qui permettrait de réfléchir de manière positive. Pour citer un exemple de difficulté administrative, la notion de soin courant, introduite dans la loi du 9 août 2004 relative à la politique de la santé publique, est délicate à utiliser par les unités de recherche clinique. En ce moment même se tient dans mon université une réunion sur la définition de cette notion et sur la manière de l'utiliser.
M. Pierre Tiberghien. Si pesants que soient les freins réglementaires ou administratifs, les scientifiques ont tous besoin que l'on réglemente et qu'on évalue l'effet de leurs découvertes sur l'usager, sur son information, sur sa sécurité virale et sur l'environnement.
Je serai plus sévère à l'égard d'un autre frein, d'ordre qualitatif celui-ci : le frein par défaut de qualité, non des recherches ou de l'innovation elles-mêmes, mais de tout ce qui relève de la préparation du dossier que les commissions évalueront. Sans doute peut-on agir en amont, en proposant d'aider le scientifique ou le clinicien, fort occupés dans leur laboratoire, à monter leur dossier et à répondre aux attentes. Ce serait d'autant plus important que l'effort de rigueur et l'accompagnement de la qualité des recherches profite à tous, particulièrement au patient.
Un dernier aspect de notre réflexion doit porter sur la qualité de l'évaluation. Dans ce domaine, on s'en remet parfois à des experts qui ne semblent pas plus éduqués que les chercheurs qui ont monté un dossier. Européaniser l'évaluation me semble un point essentiel. Ouvrir ainsi le champ réduirait les conflits d'intérêts et garantirait une expertise plus pointue.
M. Laurent Degos. Les blocages législatifs n'étaient pas les mêmes il y a quarante ans. Quand les équipes de Jean Hamburger ont pratiqué des greffes de reins à partir de personnes dont le coeur battait encore, mais qui étaient mortes cérébralement, elles ont été hors la loi pendant cinq ans. Elles ont osé cette transgression, ce que nul ne ferait aujourd'hui, car on faisait alors confiance à la science. C'est parce que ce n'est plus le cas que fleurissent les lois de bioéthique ou de protection de la personne, ainsi que les réglementations. Elles montrent qu'il faut sans doute accomplir un travail de fond en direction des populations.
Sur le plan réglementaire, nous travaillons non sur un produit fini ou sur un médicament destiné à améliorer les précédents, mais sur une innovation de rupture, qui constitue un saut technologique. Dès lors, même si l'on peut être tenté, par souci d'efficacité, de simplifier le millefeuille administratif, on est dans l'inconnu en matière de sécurité ou de protection de la personne. On se heurte à des questions plus sociétales et plus éthiques que pour tout autre produit. On mesure mal les risques. Faut-il prévoir une structure nouvelle qui évaluerait mieux le rapport entre efficacité, sécurité et protection de la personne ? Celle-ci pourrait réfléchir de manière globale et à un plus haut niveau que les instances successives que nous connaissons, dont le critère ultime est la sécurité, puisque c'est finalement l'AFSSAPS qui délivre le blanc-seing. Il serait bon de rassembler ces critères dans une vision spécifique aux sauts technologiques, tout en sachant qu'il est bien difficile de distinguer saut technologique et soin courant.
Soit on persévère dans la direction qui prévaut actuellement et qui bloque les sauts technologiques, soit on adopte une vision plus globale de l'efficacité, de la sécurité et de la protection de la personne, en se plaçant sur un plan à la fois sociétal et éthique. Puisque la société souhaite continuer dans la voie du progrès, demandons-nous si les structures actuelles nous permettent de le faire ou s'il faut adopter un dispositif plus simple, plus compréhensible et plus intégré, dans lequel la société ait sa place et qui rende possibles les sauts technologiques.
M. Jean Marimbert. Il faut en fait distinguer trois catégories : le soin courant, l'innovation et le saut technologique. Dans notre pays, on a toujours tendance à penser que les problèmes comme les solutions viennent des structures, mais je pense que les problèmes viennent surtout des processus.
Pour avoir vu pendant sept ans beaucoup de dossiers de thérapies cellulaires, je peux confirmer l'analyse de M. Tiberghien. Quand un dossier implique intrinsèquement une prise de risque, tant pour celui qui le présente que pour celui qui va l'autoriser, le groupe d'experts ou le directeur général se décide essentiellement en fonction de sa qualité. Autant dire que le système actuel ne bloque pas totalement le saut technologique.
Cependant, puisqu'on ne peut franchir l'obstacle avec un dossier trop faible, il faut prévoir un appui permettant aux chercheurs de bien monter le leur. Ils n'ont pas toujours les moyens de faire, et aucune agence régulatoire, quelle qu'elle soit, ne pourra les y aider. Cet appui est fondamental, si l'on veut aider à la prise de risque.
Pour le reste, je pense qu'on ne remplacera jamais la décision sanitaire par celle d'un groupe sociétal, même si l'on peut éclairer la décision sanitaire par la délibération et le débat collégial et sociétal.
M. le président Claude Birraux. Tout cela montre l'importance d'améliorer la gestion des interfaces.
TROISIÈME TABLE
RONDE : LES DISPOSITIFS MÉDICAUX
PRÉSIDENCE DE M.
JEAN-MICHEL DUBERNARD, MEMBRE DU COLLÈGE DE LA HAUTE
AUTORITÉ DE SANTÉ
M. le président Claude Birraux. Nous avons déjà beaucoup appris lors des deux tables rondes de ce matin. Nous allons maintenant aborder les dispositifs médicaux sous la présidence de Jean-Michel Dubernard, membre du collège de la Haute autorité de santé, qui est l'un de nos anciens - et éminents - collègues.
M. Jean-Michel Dubernard, membre du Collège de la Haute autorité de santé. Il s'agit certes, messieurs les professeurs Alain Carpentier, Ugo Amaldi et José-Alain Sahel, de nous présenter des projets innovants qui représentent des ruptures technologiques, mais surtout de nous exposer les difficultés que vous avez rencontrées.
La définition du dispositif médical est complexe, puisqu'elle recouvre tout ce qui est en contact avec l'homme. Cela va des valves cardiaques au coeur artificiel, en passant entre autres par les fauteuils roulants. C'est donc un vaste domaine, qui revêt une dimension à la fois scientifique, dont la portée peut être immense, et économique, avec un marché dominé par les petites entreprises.
Nous souhaitons discuter de toutes les étapes de votre démarche - la genèse de l'idée, la recherche de financements, les demandes d'autorisation -, qui se solde parfois, nous le savons, par l'arrêt du projet ou sa délocalisation. De ce point de vue, le domaine du dispositif médical est sans doute le plus emblématique du système.
Nous recevons donc cet après-midi trois personnalités de premier plan. Le professeur Alain Carpentier nous parlera du coeur artificiel implantable, puis le professeur Ugo Amaldi évoquera les difficultés qu'il a rencontrées dans le domaine de la protonthérapie et de l'hadronthérapie. Nous aborderons enfin, avec le professeur Sahel, les implants rétiniens - un domaine avec lequel nous nous projetons vraiment dans le futur.
La dimension internationale est très présente dans vos travaux - car les problèmes que nous rencontrons ne sont pas spécifiques à la France ; en tout état de cause, la réalité nous contraint à le prendre en compte. Lorsque M. Carpentier et moi-même étions jeunes, la France comptait vingt ou vingt-cinq entreprises de fabrication d'instruments chirurgicaux. Il n'en existe plus une seule ! De même, s'il reste trois entreprises françaises fabriquant des prothèses de hanche, leurs actionnaires sont étrangers. Au-delà de la dimension scientifique, notre débat revêt donc une dimension économique.
M. Alain Carpentier, cardiologue, hôpital européen Georges-Pompidou, président de l'Académie des sciences. Comment identifie-t-on un créneau porteur qui permettrait de faire un saut technologique ? L'avantage du chirurgien est ici d'être confronté tous les jours à des problèmes humains qu'il s'agit de résoudre. Dans mon cas, c'était le drame de voir mourir des patients de cinquante ou soixante ans, faute de solution de remplacement du coeur. Pour un médecin dont le devoir est de soigner, c'est un terrible échec. Certes, il y a toujours la solution de la transplantation cardiaque, mais seules 340 transplantations ont lieu chaque année, alors qu'il en faudrait dix fois plus.
L'insuffisance cardiaque terminale concerne 600 000 personnes chaque année, avec 80 000 nouveaux cas par an. Aux États-Unis, 500 000 nouveaux cas sont enregistrés chaque année, soit six fois plus, sans doute pour des raisons liées à l'alimentation.
Après épuisement des thérapeutiques médicales et chirurgicales d'assistance, la seule solution est le remplacement du coeur. Les besoins en ce domaine ne sont pas couverts, ni par les dispositifs connus sous le nom de « coeur artificiel » - mais qui sont en fait des dispositifs d'assistance qui, sauf exception, ne sont utilisés que dans l'attente d'une transplantation - ni par la transplantation cardiaque elle-même, en raison de contre-indications médicales et de disponibilités insuffisantes.
Il fallait donc - ce fut le point de départ du projet Carmat - un véritable coeur artificiel. Bien entendu, il ne pouvait s'agir de copier ce qui existait déjà, mais bien de faire un saut technologique - j'apprécie d'ailleurs beaucoup le titre que vous avez choisi pour cette journée d'auditions car c'est de cela qu'il s'agit ici. En ce qui me concerne, j'ai bénéficié de la relation que j'ai établie avec Jean-Luc Lagardère. Il s'est révélé un merveilleux partenaire, doté d'une vision d'avenir et de vrais moyens. Sur le plan technologique, l'aéronautique était la discipline qui pouvait répondre à nos besoins. Nous avons donc constitué avec EADS un groupement d'intérêt économique, le GIE Carmat - Carpentier-Matra -, devenu depuis la société Carmat SAS. C'est un exemple du changement de mentalité qui doit imprégner nos chercheurs : ne pas craindre de donner à leurs innovations le bénéfice de leur réalisation. J'ai vécu une époque où il était honteux en France de faire argent des applications de la science fondamentale. Comme cette éthique n'était pas celle de tout le monde, ce sont les Américains qui en profitaient. Durant trente ans, mes inventions ont ainsi été exploitées aux États-Unis.
Restait à se poser honnêtement la question de savoir si j'avais vraiment quelque chose à apporter pour résoudre les problèmes qui se posaient encore dans ce domaine. La réponse a nécessité beaucoup d'humilité - et aussi nombre de consultations pour faire le point sur ce qui existe. Nous avions dans notre jeu quelques cartes que ne possédaient pas les Américains, qui connaissent encore des difficultés avec le coeur artificiel qu'ils ont développé. Je n'en citerai qu'une : l'hémocompatibilité, c'est-à-dire le fait que le sang a tendance à coaguler au contact d'un corps étranger. J'avais une solution grâce aux bioprothèses valvulaires que j'ai inventées il y a quarante ans. Il s'agit de valves construites à partir de tissus animaux traités chimiquement pour prévenir le rejet immunologique. Elles ont l'énorme avantage de ne pas déclencher la formation de caillots contrairement aux valves en matériaux synthétiques. Avec une expérience de plus de trente ans en clinique humaine, nous avions donc une avance technologique certaine.

Présentation de M. Alain Carpentier
Mais l'idée initiale ne suffit pas toujours. En l'occurrence, nous avons constaté que si l'hémodynamique n'était pas parfaite, autrement dit si le coeur ne fonctionnait pas comme un coeur normal, on assistait néanmoins à la formation de caillots même avec ce matériau hémocompatible. Et c'est ainsi que j'ai été conduit à entrer dans l'aventure du coeur artificiel. En plus des valves de qualité biologique idéale, il me fallait en effet un coeur pourvu de qualités hémodynamiques idéales.
Quelles sont les différences de notre prothèse avec les modèles étrangers ? Tout d'abord, je l'ai dit, les matériaux bioprothétiques. Ensuite, un concept de coeur au plus près de l'anatomie et de la physiologie normales. Nombre de coeurs étrangers ont une seule pompe d'animation, alors que notre coeur en a deux avec des régulations séparées. Les courbes aortiques obtenues sont exactement celles que fournirait un coeur normal, et pourtant ce sont celles d'un coeur artificiel ! C'est vous dire le degré de perfection auquel nous sommes parvenus.
Autre avantage, l'électronique embarquée. Alors que les coeurs artificiels existants comportent une console d'animation et de régulation séparée du malade, notre coeur est « incorporé » stricto sensu, c'est-à-dire logé entièrement en lieu et place du coeur malade. Seule exception, la source d'énergie, fournie par des batteries portées à la ceinture. C'est à ce jour le seul coeur artificiel où tout est embarqué. Je rends ici un nouvel hommage à l'aéronautique. Les systèmes embarqués miniaturisés au quart de millimètre, c'est son domaine d'expertise. De même l'électronique de commande qui bénéficie des derniers progrès de la technologie.
Une deuxième avance technologique nous a été précieuse : les progrès de l'imagerie médicale. Les techniques numériques nous permettent aujourd'hui de faire des simulations - on peut par exemple incorporer virtuellement un coeur artificiel à l'intérieur d'un corps humain donné. On arrive ainsi à s'assurer que la prothèse entrera sans aucune gêne dans le corps du malade à sa place normale.
La simulation numérique nous permet aussi de voir, si le sang stagne, dans quelques zones critiques qui aboutiraient à la formation de caillots.
Autre avancée importante : la régulation médicale, copie fidèle de la régulation du coeur normal. Nous pouvons même simuler des pathologies, par exemple la réaction de la prothèse à une hémorragie virtuelle ou un accident de voiture.

Présentation de M. Alain Carpentier
Le dernier visuel que je vous montre en hommage au travail d'équipe de mon laboratoire et d'EADS, résume les progrès accomplis en dix ans de travaux acharnés.
Lorsque j'ai commencé - avec le modèle animal - le volume du coeur était de 1,250 litre. Il est passé à 0,75 litre : c'est un progrès considérable. De même, le poids est passé de 1,9 kilo à 0,9, soit le poids d'un coeur d'insuffisant cardiaque. Enfin, la consommation a été réduite de 100 à 27 watts.
Fruits de vingt ans de travail dans la plus grande discrétion, ces progrès ont aujourd'hui abouti à un produit implantable.
M. Jean-Michel Dubernard. Je vous félicite. Il semble que tout soit allé pour le mieux, mais peut-être nous parlerez-vous davantage de vos difficultés et de la concurrence internationale lors du débat.
Le professeur Amaldi, personnalité de haut niveau, italienne et savoyarde, suisse et serbe, va maintenant nous parler de la protonthérapie et de l'hadronthérapie. C'est une question sur laquelle s'est penchée la Haute autorité de santé. J'aimerais savoir exactement où nous en sommes.
J'attends aussi, Monsieur Saout, que les représentants des patients nous donnent leur avis !
M. Ugo Amaldi, physicien à l'Organisation européenne pour la recherche nucléaire (CERN). Je suis très reconnaissant à M. Birraux de m'avoir invité à vous exposer la situation et les enjeux de l'hadronthérapie française. C'est un thème qui m'est cher, car je suis associé à son développement depuis 1987, date à laquelle j'ai proposé aux professeurs Jean-Pierre Gérard et Joseph Remilleux de créer à Lyon, sur la base de ce que nous faisions au CERN et en Italie, un centre de thérapie avec ions carbone. En 2005, j'ai rencontré M. Birraux pour lui proposer la réalisation, dans celle que nous appelons à Genève « la France voisine », d'un centre de protonthérapie basé sur une technique nouvelle développée au CERN. J'expliquerai pourquoi nous n'avons pas réussi.
Par « hadronthérapie », on entend toutes les formes de radiothérapie qui utilisent des particules non élémentaires. Aujourd'hui, les protons et les ions carbones sont employés dans les centres médicaux d'hadronthérapie.
Par opposition, on qualifie de « radiations conventionnelles » les faisceaux de rayons X. Il existe en France 180 centres de radiothérapie conventionnelle publics et privés où sont traités chaque année environ 180 000 malades, soit la moitié des patients atteints d'un cancer.
Les rayons X sont produits par les électrons accélérés dans un accélérateur linéaire d'un mètre et demi de longueur, un « linac », jusqu'à 10, parfois 20 MeV.
Un linac de radiothérapie conventionnelle peut tourner autour du patient, et les angles d'irradiation peuvent être choisis de manière optimale.
La distribution de la dose en profondeur des rayons X et d'un faisceau mono-énergétique de protons est très différente. La dose déposée par un faisceau de rayons X est à son niveau maximal à quelques centimètres sous la peau, puis diminue de façon exponentielle. Beaucoup plus favorable est le comportement d'un fin pinceau de protons de 200 MeV d'énergie.
On appelle « pic de Bragg » le dépôt de dose maximum à la fin du parcours.
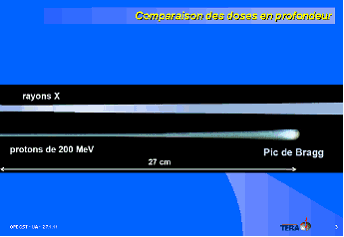
Présentation de M. Ugo Amaldi
J'en viens au cas du traitement d'une tumeur au cerveau avec les rayons X et les protons. Pour l'irradiation avec les rayons X, on utilise neuf points d'entrée. La dose intégrale distribuée l'est sur toute la masse du cerveau. Lorsque la distribution de la dose est due à quatre faisceaux de protons ayant une énergie maximale de 160 MeV, la différence se voit immédiatement : une fraction beaucoup plus petite des tissus sains est irradiée avec les protons - ce qui est essentiel pour le cerveau.
La comparaison n'est valable que parce que tous les arguments et les expériences physiques, biologiques et cliniques ont démontré que les effets des rayons X et des protons sur les cellules et les tissus sont les mêmes.
En bref, dans toutes les distributions de dose, les protons épargnent mieux les tissus sains que les rayons X, de sorte que si nous étions capables de construire un accélérateur de 200 MeV d'énergie, aussi petit que le linac pour radiothérapie conventionnelle, les rayons X ne seraient plus utilisés dans nos hôpitaux.
Malheureusement, nous n'en sommes pas là. La plupart des établissements de protonthérapie du monde ont, en leur centre, un grand « cyclotron » dans lequel les protons suivent, pendant l'accélération, une trajectoire en spirale.
Le cyclotron de la compagnie belge IBA a été installé en 2010 au centre de protonthérapie de l'Institut Curie-Orsay, en substitution du vieux synchrocyclotron construit en 1956, avec lequel les irradiations étaient faites dans deux salles avec des faisceaux horizontaux de protons. Une salle supplémentaire, équipée d'un bras isocentrique, permettra désormais de plier les faisceaux et d'irradier le patient depuis toutes les directions.
Pour utiliser de manière optimale un cyclotron et toute l'infrastructure, trois bras isocentriques sont installés dans les modèles de centre « standard » proposés par IBA.
L'investissement nécessaire pour un tel centre de protonthérapie est de l'ordre de 150 millions d'euros. Cinq entreprises peuvent produire ces systèmes. Pour une organisation privée, cet investissement devient rentable si l'on irradie plus de 1 000 patients par an, ce qui correspond à plus de 20 000 séances de traitement, puisque chaque patient bénéficie d'une séance par jour pendant cinq semaines successives. En effet, le coût de la protonthérapie par malade en Europe est aujourd'hui compris entre 20 000 et 25 000 euros, soit à peu près trois fois plus qu'une radiothérapie conformationnelle par modulation d'intensité avec rayons X, mais nettement moins que beaucoup de traitements par chimiothérapie.
Combien des 180 000 Français irradiés chaque année avec les rayons X auraient plus de probabilité de guérir et/ou d'avoir moins d'effets secondaires dans les tissus sains s'ils étaient irradiés par protons et non par rayons X ? Examinons les faits.
Dans les six dernières années, le nombre de patients traités par protons dans le monde a doublé, jusqu'à dépasser les 70 000. Cette croissance exponentielle s'accompagne d'une croissance du nombre de centres de protonthérapie, qui sont désormais plus de trente. L'an passé, les autorités sanitaires du Royaume-Uni ont décidé la construction de deux centres, l'un à Londres et l'autre à Manchester, tandis qu'aux États-Unis, la fameuse Mayo Clinic a lancé la construction de deux centres dans deux États différents.
En ce qui concerne les indications cliniques de la protonthérapie, des études médicales menées en Allemagne, en Autriche, en France et en Italie dans le cadre de la collaboration European Network for Ligh Ion Therapy (ENLIGT), coordonnée par le docteur Manjit Dosanjh du CERN, ont conclu que dans cinq à dix ans, il faudrait pouvoir traiter par protons 12 % des personnes atteintes de cancer qui relèvent aujourd'hui d'une radiothérapie conventionnelle. Pour ces patients, on prévoit des avantages en termes de taux de guérison à cinq ans et/ou de réduction des complications. Pour 1 % d'entre eux, l'irradiation avec les protons est même considérée comme nécessaire. Les cibles sont les mélanomes de la choroïde, les chordomes et les chondrosarcomes de la base du crâne, des tumeurs du massif facial et de la sphère ORL et les tumeurs pédiatriques.
En France, la protonthérapie est pratiquée dans le centre d'Orsay et dans le département de radiothérapie du centre Antoine Lacassagne à Nice. L'accélérateur « Medicyc » du centre Lacassagne, entré en fonction en 1991, est un cyclotron qui accélère les protons jusqu'à 65 MeV, énergie suffisante pour traiter les mélanomes de la choroïde.
Au centre de protonthérapie de l'Institut Curie à Orsay, les applications principales concernent les mélanomes de la choroïde, mais aussi les tumeurs intracrâniennes et les tumeurs des enfants.
J'en viens aux problèmes auxquels se heurte la réalisation de grandes installations médicales en France, avec l'exemple du projet de protonthérapie IDRA que nous avons tenté de construire en Haute-Savoie en partenariat avec le canton de Genève.
En août 2005, sur le site d'Archamps, en Haute-Savoie, j'ai présenté à M. Birraux un projet très innovant développé par le CERN, la fondation TERA, que je préside, et l'Institut national de physique nucléaire italien.
Le coeur de l'Institut pour le diagnostic et la radiothérapie avancés (IDRA) était un accélérateur linéaire de nouvelle conception, dont le prototype a été construit au CERN. Il accélère, dans une longueur de 16 mètres, les protons produits par un petit cyclotron jusqu'à l'énergie nécessaire pour la protonthérapie.
La proposition était faite par l'entreprise française Advanced Accelerator Applications (AAA). Son président-directeur général, M. Stefano Buono, est un ancien du CERN, qui a construit un centre de production d'isotopes à Saint-Genis.
Dans ce projet, auquel était également associé M. Adolfo Fucci du CERN ici présent, TERA aurait eu la responsabilité du linac et AAA du cyclotron et de la production des isotopes employés dans le diagnostic des tumeurs et leur thérapie. L'idée était de construire le linac et les aimants pour le transport de faisceaux de protons avec des entreprises françaises.
En 2005 et 2006, M. Birraux a pris des contacts - à haut niveau - avec les autorités de santé régionales et le canton de Genève. Mais le projet n'a pu se réaliser, notamment parce qu'il était question de construire à Lyon le centre ETOILE, dédié à la radiothérapie par ions carbone que j'évoquerai. On a, hélas, négligé notre principal argument : la thérapie avec protons concerne un grand nombre de patients, tandis que la thérapie avec ions carbone est efficace sur les tumeurs radiorésistantes.
Je regrette encore cette occasion manquée d'une collaboration étroite entre la France et mon pays dans le cadre du CERN.
Combien de salles de protonthérapie faudrait-il avoir en France dans cinq ans ?
Pour prendre en charge 1 % de toutes les tumeurs aujourd'hui irradiées avec les rayons X, il en faudrait au moins cinq. Les deux centres existants ne suffisent donc pas, et IDRA n'a pu voir le jour. Heureusement, il y a deux nouveaux projets. Dans le cadre du projet de cancéropôle de Toulouse a été proposée la construction d'un centre de radiothérapie avancée, avec un seul faisceau horizontal de protons. Le projet d'Institut de physique médicale dédiée aux traitements des cancers (IMPACT), qui associe notamment le centre Antoine Lacassagne, sera dédié au développement des nouvelles techniques de traitement des tumeurs profondes par les protons et les radioéléments guidés par la thérapie. Le cyclotron supraconducteur pour la protonthérapie est le coeur d'un contrat AIMA - IBA signé en juillet 2010.
Bien que le recrutement des patients ne soit pas organisé, ces deux projets placent la France en bonne position pour les années à venir.
J'en viens à la thérapie par ions carbone. Nous avons créé en 1992, pour la conception et la construction de centres avec ions carbone, la fondation italienne TERA. J'ai également lancé au CERN le Proton ion medical machine study (PIMMS). Nous avons, enfin, obtenu en 2002 le premier financement du Centro nazionale di adroterapia oncologica (CNAO), construit à Pavie - qui a coûté 125 millions d'euros, dont 80 % financés par le ministère de la santé italien. La première irradiation y aura lieu dans environ six mois.
Un projet similaire lancé en Allemagne par le professeur Gerhard Kraft a permis de construire le synchrotron du centre de Heidelberg (HIT). Siemens Medical est en train d'achever la construction de deux autres centres d'ions carbone à Marburg et à Kiel.
De leur côté, nos collègues autrichiens ont obtenu il y a deux ans l'approbation de leur projet MedAustron. Ils ont reçu de la fondation CNAO les plans d'exécution de notre centre - ce que nous aimerions voir arriver aussi pour le centre ETOILE de Lyon.
Pourquoi des ions carbone ? Tout simplement parce qu'ils ont une grande capacité à ioniser les molécules qui sont au centre des cellules. Ils permettent ainsi de traiter les 3 % de malades irradiés avec rayons X qui ont des tumeurs radiorésistantes - soit, en France, 5 000 patients par an. Deux projets sont d'ailleurs en cours : ETOILE à Lyon et ARCHADE à Caen.
Pour conclure, on peut dire que les enjeux de la protonthérapie et de l'hadronthérapie sont différents. La protonthérapie a vocation à remplacer une grande partie, sinon toute la radiothérapie par rayons X, alors que l'hadronthérapie est une niche.
La France est peu équipée pour la protonthérapie, mais si les projets de Toulouse et de Nice démarrent, ils lui permettront de rattraper une partie de son retard.
En ce qui concerne l'hadronthérapie, il faut accélérer le projet ETOILE, qui permettra à la France d'être bien placée, même si elle reste en retard par rapport à l'Allemagne et à l'Italie.
M. Jean-Michel Dubernard. Dans ce domaine, il faut être international et européen.
Le projet ETOILE a été lancé en 1997. Comment se fait-il qu'en 2011, on doive repartir à zéro - ou presque ? Il y a là une vraie interrogation. J'espère que les représentants d'institutions qui sont présents pourront nous aider à y répondre tout à l'heure.
Je donne maintenant la parole à José-Alain Sahel, célèbre ophtalmologue de l'hôpital des Quinze-Vingts, qui va nous parler des implants rétiniens. C'est pour nous, à la Haute autorité de santé, le dispositif qui anticipe le plus sur l'avenir.
M. José-Alain Sahel, directeur de l'Institut de la vision. Je vais donc vous présenter l'état d'avancement des recherches sur la rétine artificielle, en précisant d'emblée que les perspectives de développement sont encore considérables.
L'oeil fonctionne de manière schématique, comme une caméra. La cornée laisse passer les images, avec le cristallin. Elles sont focalisées sur une pellicule sensible de 300 microns d'épaisseur.
La rétine est un tissu composé de milliards de neurones, qui ressemble à une pellicule photo : elle capte de la lumière pour la transformer en signal, mais elle traite le signal - forme, mouvements, contraste, couleur... Le signal quitte l'oeil par le nerf optique pour atteindre le cerveau, qui est le véritable siège de la vision - l'oeil ne servant que de premier étage, mais appartenant au cerveau.
Dans L'oeil et l'esprit , Maurice Merleau-Ponty écrit que l'oeil ressemble à un computeur du monde. C'est un système qui traite l'information, qui détecte la lumière, qui utilise le contraste spatial, puis temporel, pour coder formes, mouvements et couleurs.
Le signal qui quitte l'oeil pour aller vers le cerveau est un signal déjà complexe et traité. Lorsque j'ai commencé l'ophtalmologie, mon chef de service disait que l'oeil n'était qu'une partie du cerveau et qu'on ne pourrait jamais remplacer la rétine. Il avait heureusement tort.
Néanmoins, on ne peut pas remplacer toute la rétine. Cette présentation a donc pour objet l'utilisation des cellules restantes dans la rétine pour les stimuler et générer une sensation lumineuse.
Nous sommes aujourd'hui confrontés à des maladies qui sont responsables d'un nombre important de malvoyances et de cécités. Si l'on excepte le diabète, les deux principales sont la dégénérescence maculaire liée à l'âge (DMLA) - qui touche en France 1,5 million de personnes, dont 20 % sont légalement considérées comme aveugles -, et les maladies génétiques comme la rétinopathie pigmentaire, qui peuvent frapper dès l'enfance ou l'adolescence et sont la première cause de cécité et de malvoyance avant cinquante ans. Dans ces deux maladies, les photorécepteurs - qui constituent le premier étage de la vision et reçoivent normalement la lumière pour la transformer en signal - disparaissent. Mais le réseau de neurones qui transmet l'information au cerveau, lui, existe encore. L'objectif de la rétine artificielle est donc de stimuler ces neurones en réponse à la lumière, pour déclencher des sensations lumineuses. Idéalement, il s'agit de restaurer deux fonctions principales pour retrouver une autonomie : la lecture et la mobilité.
Restaurer la mobilité, qui est notre premier objectif, implique de restaurer une capacité de détection de la lumière et une capacité de discrimination. Je vous parle aujourd'hui de la rétine artificielle, mais il faut savoir qu'il existe d'autres approches, comme les cellules souches ou la thérapie génique, qui sont très prometteuses. Si j'en avais le temps, je vous décrirais une approche qui combine les bioprothèses et la thérapie génique, l'optogénétique. Je vais cependant cibler mon propos, comme prévu, sur la prothèse électronique.
Il s'agit donc, je l'ai dit, de stimuler la rétine restante. On a tenté les implants sur la rétine, sur le nerf optique ou directement sur le cerveau. En ce qui concerne le cerveau, un programme est en cours avec le professeur Benabid et un autre à Salt Lake City. Cela peut représenter une possibilité pour des patients qui ne sont pas accessibles aux autres thérapeutiques ; mais tant qu'existe une capacité à traiter le signal, il est logique de l'exploiter et donc de stimuler les cellules restantes. Quant à la stimulation du nerf optique, elle est tentée à Louvain depuis quelques années, mais les résultats sont très médiocres, car tous les neurones sont compactés et la sélectivité de la stimulation en termes topographiques est très mauvaise.
Nous nous sommes donc concentrés sur la stimulation de la rétine. Le système que nous avons testé est la deuxième génération d'implants. Les États-Unis, en l'occurrence le département de la défense, ont investi des centaines de millions de dollars dans cette technologie ; l'Allemagne en avait investi des dizaines, dans le cadre d'un plan stratégique pour l'industrie, dans les années 1990. Il n'est donc pas étonnant que les pionniers en ce domaine se trouvent dans ces derniers pays.
En France, l'argent commence seulement à arriver - il aura fallu dix ou quinze ans - sur un projet qui sera la troisième génération.
Je vais maintenant vous décrire les essais cliniques que nous avons menés avec nos compétiteurs allemands et américains au centre des Quinze-Vingts. Il s'agit de l'Argus II, essai mené dans une dizaine de centres - quatre en Europe et cinq aux États-Unis. Je précise qu'il n'y a aucun conflit d'intérêts.
L'étude est conduite sur trois ans. Le premier malade a été implanté aux Quinze-Vingts le 4 février 2008. Le suivi porte sur la fonction visuelle, l'objectif étant d'assurer la sécurité et d'évaluer la qualité de vie. La population est composée de patients aveugles depuis une quinzaine d'années en raison d'une rétinopathie pigmentaire. Il fallait cependant vérifier que persistait une capacité à stimuler le nerf optique, en déclenchant par de petits courants électriques une sensation lumineuse. Ces patients n'étaient accessibles à aucun autre traitement. Sur une trentaine de patients implantés, un seul - aux États-Unis - a dû subir une ablation de l'implant en raison d'un problème de cicatrisation.
Le système est le suivant : une caméra posée sur les lunettes transmet les informations à un processeur externe - tout le traitement de l'information se fait en externe, ce qui permet de faire évoluer le système. Le processeur transmet ensuite l'information à un émetteur posé sur les lunettes, lequel les transfère, par l'intermédiaire d'un câble, à une antenne suturée sur l'oeil. Le système est donc implanté sur la rétine. Un de nos patients avait plus de soixante électrodes implantées sur la rétine, le système de fixation consistant en un tout petit clou. Ces soixante électrodes, au contact des cellules qui forment le nerf optique, déclenchent des sensations lumineuses.
S'agissant des effets indésirables, le taux de complications est de l'ordre de 5 %, aucun patient n'ayant perdu son oeil et tous connaissant - à des degrés variables - une amélioration de leur situation.
Ainsi, 97 % des patients sont capables de localiser une tache lumineuse sur un écran, et plus de la moitié de dire comment bouge une ligne sur cet écran. Une partie d'entre eux est capable de voir des barres obliques, et environ 20 % de lire des lettres et des mots - ce qui n'était pas prévu au départ.
Les premiers tests étaient des tests de mobilité - trouver une porte, une fenêtre, suivre une ligne blanche... La majorité des patients est capable de retrouver une porte ou de suivre une ligne. Pour les lettres, le résultat est assez spectaculaire.
Mais le plus important sera la rééducation, mois après mois, du cerveau pour apprendre à utiliser ces informations. C'est un deuxième défi, puisqu'une prothèse de ce type ne pourra pas fonctionner sans protocoles, sans ergothérapeutes et sans professionnels de la rééducation. La coopération avec le patient est donc extrêmement importante.
Nous avons réalisé un petit film sur les performances des patients. Les tâches sont les suivantes : 100 % de perceptions lumineuses, 96 % de localisations, 57 % de directions et mouvements. Un quart des patients ont une acuité visuelle, et 50 % identifient une lettre.
Les malades emmènent le système chez eux. Certains l'utilisent en permanence, grâce à des batteries, d'autres deux heures par jour - ceci pour localiser des objets, s'orienter, se déplacer, détecter une source lumineuse, effectuer certaines tâches ou même lire - avec de gros caractères. Certains patients sont en effet capables de lire les plus grosses lettres du test d'acuité visuelle classique.
Nous avons essayé d'évaluer la qualité de vie, avec une étude qui n'avait pas été prévue à l'origine. On peut regrouper les tests en tâches ou en domaines. En tâches élémentaires, les résultats sont hétérogènes ; en certains domaines, ils sont relativement homogènes. Les tâches élémentaires sont, par exemple, de manger tout seul, d'aller aux toilettes, de reconnaître quelqu'un dans la rue... Lorsqu'on regarde l'ensemble des tâches en faisant la distinction entre tâches visuelles pures, tâches visio-motrices et tâches motrices, on constate une amélioration qui est dans une échelle logarithmique et qui représente l'équivalent du meilleur bénéfice thérapeutique observé aujourd'hui dans la DMLA, à savoir un quasi-triplement des capacités. Rappelons-nous qu'on part d'une cécité totale !
Le marquage CE pour la mise sur le marché est officieusement accordé. La demande d'autorisation a été déposée en Allemagne, en Suisse, en Grande-Bretagne. Elle le sera bientôt en France. Je suis également en pourparlers avec l'assurance maladie, puisque la partie réhabilitation sera aussi importante que la partie chirurgicale.
Il est important de ne pas avoir trop de centres, car une chirurgie de ce type doit être parfaitement maîtrisée. Il ne s'agit pas de vendre beaucoup de systèmes, mais de les valider.
Les personnes concernées seront des aveugles de plus de vingt-cinq ans, atteints de dégénérescences très profondes, avec peut-être des performances visuelles un peu meilleures. Il faut que le patient ait vu dans l'enfance pour que le cerveau visuel soit capable d'interpréter l'information. Si on n'a pas vu avant, le système visuel utilise d'autres sensorialités - audition, toucher.
L'enjeu de l'avenir, c'est la résolution. Nous avons aujourd'hui 60 électrodes. Nous avons travaillé sur les capacités du cerveau à traiter l'information, et nous sommes arrivés à la conclusion qu'avec 600 pixels, on est capable de reconnaître un visage, surtout si l'on bénéficie d'une échelle de gris. Avec 600 pixels et vingt séances de rééducation, on peut reconnaître 80 % des mots, et avec 60 pixels seulement, on est capable de reconnaître quelques mots.
Comment augmenter la résolution ? Nous travaillons actuellement, dans le cadre d'un programme européen et ANR avec le CEA, avec une école d'ingénieurs - qui a remporté l'an dernier le prix de l'Ingénieur de l'Usine nouvelle - pour développer de nouvelles méthodes. Les brevets sont français, mais ils ne seront peut-être pas exploités en France. Il s'agit en particulier de techniques de couverture carbone, avec du diamant, qui permettent d'améliorer la biocompatibilité. Je rappelle en effet que le carbone est biocompatible.
Une autre approche consiste à utiliser des électrodes tridimensionnelles, car elles sont davantage au contact des neurones et génèrent des courants intermédiaires.
La vision, c'est 90 % des informations qui nous viennent du monde extérieur. L'audition apporte la communication avec les autres, la vision apporte la communication avec le monde. Elle dépend de la relation entre sensibilité, sensorialité et intellectualité. Nous ne sommes pas capables de reconstituer tout cela, mais la chirurgie essaye d'en restaurer une partie. C'est un long parcours.
DÉBAT
M. Jean-Michel Dubernard. Ces trois interventions nous donnent l'impression que tout se passe pour le mieux. Mais s'agissant des implants rétiniens, je suis tout de même sidéré que l'on n'arrive pas à faire des essais européens communs. Je l'ai personnellement constaté en poussant l'idée au niveau de la Haute autorité de santé, même si nous avons réussi en décembre - du moins je l'espère - à lancer une démarche avec les implants cochléaires. Nous avons pourtant besoin de coopération européenne dans ce domaine !
Je suis également frappé que vous ne proposiez pas de méthodologie d'essais cliniques. Je m'adresse en particulier à M. Amaldi : nous n'avons rien de vraiment démontré, sur le plan méthodologique, en termes de résultats cliniques.
Quel type d'obstacles rencontrez-vous ? Pour M. Carpentier, tout va bien... mais il y a tout de même un problème de concurrence. Pour M. Amaldi, il s'agit davantage d'un problème « politique » - au sens le moins noble du terme. Pour M. Sahel, enfin, c'est le problème de la coopération européenne. Parlez-nous des difficultés que vous rencontrez !
M. le président Claude Birraux. Dans le cas de M. Amaldi, je me demande si le complexe du not inventing here n'a pas joué chez les autorités qui ont été sollicitées. Pour ETOILE, dont on parle depuis 1997, on repart à zéro.
M. Jean-Michel Dubernard. Puissent les représentants de la direction générale de la santé (DGS) et de la direction générale de la sécurité sociale (DGSS) ici présents nous entendre ! Que s'est-il exactement passé ? Je sais qu'il y a eu une « tension » entre Lyon et Caen et qu'il a finalement été décidé qu'on ferait la recherche à Caen et la clinique à Lyon, mais il faut reconnaître qu'on ne sait plus vraiment où on en est...
M. Ugo Amaldi. Je vous ai dit qu'il fallait distinguer la protonthérapie et la thérapie avec ions carbone. La protonthérapie se compare aux rayons X, car la physique, la biologie et la clinique nous disent que les effets sont les mêmes. Votre argument n'est pas juste, car lorsqu'on a fait des progrès dans la thérapie avec rayons X, moyennant des frais importants, on n'a jamais fait d'essais cliniques de phase II ou III : il était en effet évident que mettre plus de dose dans la tumeur et moins dans les tissus sains était préférable. Les protons, c'est cela. C'est pourquoi de grands thérapeutes disent qu'il serait contraire à l'éthique de comparer les rayons X et les protons.
Pour les ions carbones, il existe en revanche une différence dans la qualité de radiation. Nous procédons donc à des études de phase III dans le cadre européen. Il s'agit de faire des comparaisons avec les protons - et non les rayons X.
M. Jean-Michel Dubernard. C'est fort bien, mais il n'y a pas de véritables données scientifiques.
M. Ugo Amaldi. Il n'y en a pas besoin : on a bien acheté des appareils de rayons X sans faire d'études.
M. Jean-Michel Dubernard. C'est une erreur de raisonnement - et je n'hésite pas à le dire devant Claude Birraux, car je pense que c'est l'une des explications au retard qui a été pris en France.
M. Jean de Kervasdoué, ancien directeur général des hôpitaux, membre de l'Académie des technologies, professeur au Conservatoire national des arts et métiers. Je proposerai une explication administrative de cette question, qui me passionne depuis longtemps - j'ai en effet autorisé la neutronthérapie à Nice, en 1985, alors que j'étais administrateur de la filiale du CEA.
Les hauts fonctionnaires, notamment ceux de la sécurité sociale, ont pour seul raisonnement celui de la règle de trois : quand un dispositif coûte cher en valeur absolue, il faut éviter de l'installer. C'est la raison pour laquelle, alors que pratiquement tous les hôpitaux français ont été rénovés, ceux qui accusent le plus grand retard sont les hôpitaux de Paris, Lyon et Marseille.
Par ailleurs - et cette raison est peut-être plus profonde -, alors que l'administration est habituée à discuter avec l'industrie pharmaceutique qui, puissante et organisée, paie le plus souvent les démarches administratives, l'industrie du matériel biomédical est essentiellement constituée de PME. Dans le cas que nous évoquons, des chercheurs du CERN peuvent fabriquer une machine d'essai dont deux petites sociétés fabriqueront des parties mais, dans un secteur où il n'existe aucune entreprise française comparable, à Siemens,par exemple, on ne trouve pas de porteur de projet.
Ainsi, le coût d'un éventuel tarif de remboursement n'a pas encore fait l'objet de discussions avec l'assurance maladie et les malades sont envoyés en Allemagne, de sorte que la France manque d'expérience en la matière.
Il faut absolument que l'administration accepte de tester des machines coûteuses. Avec un coût annuel de l'ordre de 30 000 euros, le traitement est beaucoup moins cher que de nombreux médicaments ou qu'une dialyse dans un centre spécialisé - laquelle coûte environ 80 000 euros par an.
Or, si rien n'a changé depuis le départ de la personne qui était jadis responsable des technologies biomédicales à la direction générale de la santé, personne n'est chargé de ce dossier au sein de l'administration.
Un article publié dans The Lancet voilà quelques mois indiquait que le traitement radiothérapeutique du cancer du sein avait des résultats analogues à ceux de 30 ou 35 séances de radiothérapie. Or, personne ne porte les « innovations immatérielles » de ce projet, qui se heurte du reste à l'opposition des radiothérapeutes.
Il importe donc que le ministère de la santé - plutôt que les instances de régulation - assure un suivi de ces innovations thérapeutiques. Le cas du professeur Carpentier, qui a surmonté les difficultés grâce à sa rencontre avec un partenaire aussi déterminé que lui-même, est plutôt l'exception que la règle.
Ayant été patron de PME et ayant créé un club consacré à ces questions, je sais par expérience qu'il faut, pour faire avancer un projet, prendre contact avec cinq ou six instances - ministère de la santé, direction de la sécurité sociale, assurance maladie, ministère de l'industrie, ministère de la recherche... Un guichet unique pour les innovations thérapeutiques serait bienvenu. Le tarif interministériel des prestations sanitaires (TIPS), qui est l'acte de remboursement de l'assurance maladie, est souvent défini brutalement, sans prendre en compte les conséquences industrielles.
Le problème ne tient donc pas tant à la technologie - qui mériterait d'être essayée, pour un coût de 150 à 200 millions d'euros - qu'à la sociologie administrative.
M. Jean-Michel Dubernard. Je souscris totalement.
Mme Chrystelle Gastaldi-Ménager, représentante de la Direction de la sécurité sociale. Je précise que, venue entendre les débats, je n'ai pas de mandat pour m'exprimer au nom de la Direction de la sécurité sociale (DSS). Ces innovations potentielles n'ont pas encore fait l'objet des démonstrations cliniques indispensables préalablement à la décision de prise en charge par la Direction. J'entends bien qu'une coordination entre les différents intervenants serait souhaitable, mais la DSS n'est pas responsable de l'ensemble de difficultés que rencontrent les innovateurs.
M. Jean-Michel Dubernard. Merci par avance, madame, de faire remonter le message auprès de la DSS.
M. Bernard Avouac, rhumatologue, représentant du Syndicat national de l'industrie des technologies médicales (SNITEM). Ne confondons pas « démontrer » et « mesurer ». Le professeur Amaldi a bien expliqué que, pour lui, l'utilité de la protonthérapie ne fait aucun doute, car elle concentre l'effet thérapeutique là où il doit s'exercer, au lieu de le disperser là où il ne doit pas le faire. S'il faut attendre de pouvoir mesurer comparativement les résultats - ce qui est certes nécessaire pour tarifer ces soins - pour conclure qu'il y a innovation, on prend un retard considérable. Il faut prendre les décisions plus tôt.
M. Jean-Michel Dubernard. Je le répète, un de mes confrères m'a déclaré que, s'il était atteint de l'une des six tumeurs qu'il m'a énumérées, il irait sans hésitation se faire soigner au Japon. Je le répète aussi, nous manquons de preuves absolues.
M. José-Alain Sahel. J'ai traité des centaines de patients souffrant de mélanomes oculaires. Beaucoup d'yeux ont été perdus et de nombreux patients ont été énucléés, puis le cobalt, que la France a utilisé longtemps, a eu des résultats catastrophiques. Une étude des National Institutes of Health lancée en 1986 aux États-Unis a comparé pendant quinze ans, sur des centaines de malades, le traitement à l'iode 125 et la protonthérapie. Elle a montré avec une grande évidence que la précision du pic de Bragg permettait des résultats incomparables et avait changé les pratiques. Si les programmes hospitaliers de recherche clinique (PHRC) n'étaient pas ce qu'ils sont malheureusement devenus, il serait possible, pour un certain nombre de thérapeutiques, de mener des études comparant les différentes approches sans dépendre de l'industrie. Il s'agit là d'une difficulté qui dépasse largement celle du « device » (dispositif) proprement dit.
L'assurance maladie devrait être consultée et financer les différentes approches. Un exemple cruel : dans six mois, nous connaîtrons les résultats d'une étude menée aux États-Unis sur la dégénérescence maculaire liée à l'âge. Lorsque nous avions demandé à lancer une telle étude en France, on nous avait répondu qu'il appartenait à l'industrie de la financer, mais nous savions pertinemment que, bien que notre projet ait obtenu la note de 12 sur 12 à l'évaluation scientifique, l'industrie ne le financerait jamais, parce que le médicament que nous voulions étudier coûtait vingt fois moins cher que le médicament de comparaison.
J'en viens au problème des devices . L'Europe dispose de cet avantage apparent que, lorsque le marquage « CE » est obtenu, le problème n'est plus que financier et de nouvelles autorisations ne sont plus nécessaires, ce qui nous donne, pour l'utilisation de certains systèmes - implants rétiniens ou autres techniques -, une avance de deux ou trois ans par rapport aux États-Unis. Ce mécanisme n'est cependant pas une garantie de sécurité. Ainsi, le marquage CE ne garantit pas qu'un implant cristallinien sera indiqué pour telle ou telle pathologie. Le bénéfice rendu des technologies n'est pas évalué en aval de leur mise sur le marché ou de leur utilisation sauvage.
L'étude relative à la rétine artificielle porte sur 30 aveugles qui voient à nouveau. On pourrait lui reprocher de ne pas porter plutôt sur 300, ou de suivre un protocole qui ne cesse d'évoluer. Or, s'il n'évoluait pas, ce devrait être un signe de génie ou de rigidité. Surtout, il faut garder à l'esprit que ce qui nous importe est de mesurer un bénéfice, notamment en termes de qualité de vie. J'ignore ce qu'il faut pour convaincre la sécurité sociale de l'utilité de ce dispositif et un dialogue devrait s'engager très tôt, lors de la conception des études.
Enfin, si l'innovation commence parfois dans nos laboratoires, elle se termine souvent ailleurs, car nous ne disposons pas des systèmes nécessaires pour les faire aboutir. Le Pr. Carpentier a bien expliqué à ce propos que le succès de son projet tient à sa ténacité et à la chance d'une rencontre.
M. Jean-Michel Dubernard. Permettez-moi de continuer à tenir mon rôle de provocateur. La Commission nationale d'évaluation des dispositifs médicaux et des technologies de santé (CNEDiMTS) mène une réflexion sur ces questions et Alain Bernard, qui en est vice-président, étudie l'opportunité de méthodologies particulières pour les dispositifs médicaux, qui ne sont pas des médicaments. La question se pose aussi pour les thérapies cellulaires, qui ont elles aussi besoin d'un cadre particulier.
M. Christian Saout, président du Collectif interassociatif sur la santé (CISS). Ce matin, j'aurais voulu demander à M. Jean Marimbert (Afssaps) quelles étaient les difficultés réglementaires ou éthiques rencontrées. Si les sauts technologiques ou innovations dans le domaine de la santé constituent un bloc, il faut créer une enceinte d'arbitrage qui leur soit propre, distincte des organes de régulation. Cette enceinte, qui pourrait reposer sur des instances existantes et être composée de personnes désignées par les parlementaires à une majorité des deux tiers, serait capable de dégager, au fur et à mesure des besoins, des règles de jurisprudence administrative.
La situation actuelle ne peut pas durer. Les patients attendent des solutions. Des situations exceptionnelles appellent des solutions exceptionnelles, comme on l'a vu à propos du VIH, pour lequel on a créé des autorisations temporaires d'utilisation qui n'existaient pas précédemment. Il est temps de créer un régime spécifique des innovations en santé, qui aurait « ses règles propres et ses contingences particulières », selon une formule du Conseil d'État.
Malheureusement, chaque fois que je viens exposer une idée dans l'enceinte du Parlement, vous en faites autre chose.
M. Jean-Michel Dubernard. Monsieur Carpentier, est-il vrai que des concurrents seraient sur le point d'implanter sur l'homme un autre dispositif de coeur artificiel ?
M. Alain Carpentier. Pour le coeur artificiel, les problèmes se posent très en amont de ceux qui sont évoqués cet après-midi. En effet, aucune comparaison statistique n'est possible, car il n'existe aucune alternative : faute de substitut cardiaque efficace, le patient n'a pas d'autre perspective que la mort. C'est pour nous un argument, notamment devant le Comité consultatif national d'éthique.
Le coût important de l'appareil nous place cependant devant un problème moral. J'avais déclaré à Jean-Luc Lagardère que nous ne ferions le coeur artificiel que si son prix ne dépassait pas celui d'une transplantation cardiaque, et j'ai inscrit cette condition dans le cahier des charges. Il est du reste rare de poser le problème financier avant le problème scientifique mais, à mon avis, il importe que les médecins, souvent taxés d'être dépensiers, intègrent dans leur réflexion les conditions économiques et sociales de leurs réalisations. Le problème moral est d'autant plus aigu que la situation financière que connaît notre pays est préoccupante avec une dette qui ne cesse de croître aux dépens de nos enfants. On doit brutalement poser la question : cela a-t-il un sens d'implanter à une personne un appareil qui coûte 250 000 euros ?
M. Jean-Michel Dubernard. Et demain ?
M. Alain Carpentier. C'est simple : ça marche ou ça ne marche pas. « Marcher » veut dire notamment que le malade gagne en qualité de vie, et n'est pas astreint à être relié à une console de commande. Si ça marche, nous passerons à l'échelle mondiale : le succès se verra quand les étrangers viendront se faire traiter en France.
M. Jean-Michel Dubernard. Quelles sont les prochaines étapes à franchir ?
M. Alain Carpentier. La première implantation sur l'homme, avec six premiers cas, devrait intervenir dans l'année.
M. Jean-Michel Dubernard. Est-il vrai qu'une expérience similaire est en cours en Italie ?
M. Alain Carpentier. Il y en a dans le monde entier, mais il s'agit de faux coeurs artificiels car, comme je l'ai expliqué, ils sont reliés à une console et sont utilisés à titre temporaire le plus souvent, ce qui n'offre pas la même qualité de vie. Nous ne jouons pas sur le même terrain.
M. Jean-Michel Dubernard. Avons-nous pris du retard et aurions-nous pu aller plus vite ?
M. Alain Carpentier. Il me semble au contraire que nous avançons. Si ça réussit, nous serons très en avance. Rien n'est gagné, mais nous avons une chance sur deux de réussir.
M. le président Claude Birraux. Pour ce qui concerne l'hadronthérapie, les études cliniques menées aux États-Unis sont-elles suffisantes pour que les autorités sanitaires françaises agréent un appareillage, ou faut-il à nouveau faire la preuve de l'efficacité du dispositif ?
M. Jean de Kervasdoué. Si les dossiers extérieurs sont de très bonne qualité, il n'y a pas de souci.
M. Jean-Michel Dubernard. La population cible est très faible.
M. Jean de Kervasdoué. Il y aurait assez de cas en France pour justifier une machine.
M. Jean-Michel Dubernard. Les chiffres ne sont pas convaincants.
M. Ugo Amaldi. Le terme d'« hadronthérapie » a un sens collectif : il faut distinguer les protons et les ions. L'hadronthérapie pratiquée au centre Étoile ou à Pavie et Heidelberg, qui utilise les ions, est parvenue au niveau des tests de phase 3. Quant à la protonthérapie, de nombreuses preuves démontrent qu'elle est plus efficace pour certaines thérapies, notamment pour l'oeil.
M. Jean-Michel Dubernard. L'efficacité n'est pas prouvée.
M. Ugo Amaldi. Pour les protons, la phase 3 n'est pas nécessaire.
M. Christian Saout. Monsieur le professeur Carpentier, lorsque vous évoquez une probabilité d'une chance sur deux, à quoi tiennent les risques d'échec ? À la technique ou aux procédures administratives ?
M. Alain Carpentier. Cela n'a rien à voir avec les procédures administratives mais avec la réaction inconnue de ce coeur avec le corps humain. Sur banc d'essai, ce coeur artificiel fonctionnera au moins cinq ans sans panne, conformément au cahier des charges, mais la réaction biologique malade-machine est la grande inconnue.
M. Christian Espagno, neurochirurgien, membre de la CNEDIMTS. En France, le problème du financement du début du passage à la phase clinique n'a toujours pas été résolu. À l'exception de situations telles que celle du professeur Carpentier, il faut recourir à des mécanismes dont l'insuffisance est largement reconnue, ou trouver des financements ouvrant à tous, dans toutes les conditions possibles, l'utilisation de ces dispositifs. Dans l'attente de dispositions législatives et réglementaires appropriées, la situation est aujourd'hui manichéenne : s'il existe des arguments forts, une thérapie est ouverte à tout le monde ; dans le cas contraire, à personne.
M. Jean de Kervasdoué. Les innovateurs sont en effet placés dans une situation impossible : l'expérimentation de phase 3 est exigée, mais il n'y a pas de financements pour la réaliser.
M. José-Alain Sahel. Si on n'est pas capable de participer à des expérimentations de phase 2, on ne peut pas revisiter les prises en charge courantes des malades, qui doivent pourtant toujours être confrontées à l'innovation.
Par ailleurs, notre pays, qui s'enorgueillit de ses magnifiques écoles d'ingénieurs, a laissé la CGE disparaître et nous en payons encore les conséquences. Si les innovateurs, les ingénieurs biomédicaux français ne peuvent pas développer leurs innovations parce que le système ne le leur permet pas, et qu'elles sont développées ailleurs, les conséquences sont à la fois de santé publique et économiques.
M. Jean-Michel Dubernard. Si je n'étais tenu à une certaine réserve, j'aurais bien des choses à dire à propos des greffes. Par ailleurs, les avis que rend la Haute autorité de santé pour aider les décideurs à décider doivent être fondés sur des arguments solides.
M. Laurent Degos, ancien président de la Haute autorité de santé. La difficulté du passage de la recherche à la clinique, déjà évoquée ce matin, est encore plus visible pour les dispositifs présentés cet après-midi. De fait, il n'existe pas d'instance ni de méthodologie européenne pour les dispositifs. En outre, ces appareils coûtent très cher et les essais de recherche sont donc eux aussi très coûteux. Faut-il des autorisations, et qui doit les donner ? Faute d'un Lagardère, d'où vient l'investissement ? Pour décider de rembourser un acte médical plus cher que les actes usuels, l'assurance maladie a besoin de preuves cliniques et doit savoir qu'il existe déjà des appareils. Il s'agit là d'une situation de blocage.
Ainsi, dans le cas de l'hadronthérapie, s'est d'emblée posée la question du prix de l'acte, avant même de disposer de l'appareillage et de preuves cliniques, ce qui ne permettait pas à l'assurance maladie de prendre une décision de remboursement. Les dispositifs médicaux sont donc entourés d'un plus grand flou encore que la thérapie cellulaire et le médicament. Sans doute faudrait-il, comme le proposait M. Saout, créer un lieu de réflexion, un guichet unique permettant de prendre en compte tous les éléments de risque et de bénéfice. Compte tenu de leur potentiel pour la collectivité, il faut sortir les sauts technologiques, les innovations de rupture, du lot commun soumis aux régulations usuelles.
M. Alain Carpentier. Il est très important d'évaluer le coût de l'acte, mais il faut aussi évaluer l'absence d'acte. L'insuffisance cardiaque traitée médicalement à l'hôpital coûte trois fois plus cher que le coeur artificiel : cela doit être intégré dans l'équation.
M. Jean-Michel Dubernard. De cette journée intéressante, je retiendrai trois choses.
Tout d'abord, les trois séances ont fait apparaître la dimension de coopération internationale, notamment européenne, qui se dégage dans les domaines évoqués.
Ensuite, quelques pistes intéressantes ont été esquissées quant au rôle des agences de régulation - même si la question de l'expertise n'a pas été abordée. À côté des conflits d'intérêts financiers, relativement faciles à régler, les conflits d'intérêts intellectuels sont plus complexes. Quant au principe de précaution, présenté par M. de Kervasdoué, la version inversée qu'en a exposée M. Avouac m'a paru d'un grand intérêt.
Enfin, il faut défendre le Parlement, attaqué ce matin par certains intervenants, car il représente la société. Ceux qui pensent différemment doivent faire passer leurs messages progressivement. C'est du reste ainsi, par étapes, que procède la bioéthique, ce qui me semble justifier le principe d'un réexamen quinquennal de la législation en la matière.
DISCOURS DE CLÔTURE DE M. CLAUDE BIRRAUX, DÉPUTÉ, PRÉSIDENT DE L'OPECST
M. le président Claude Birraux . Nous ne sommes que le Parlement, pas l'exécutif.
Au terme de cette journée très riche et savante, je voudrais exprimer mes plus vifs remerciements aux orateurs, aux participants et aux membres du panel d'experts. La remarquable qualité des propos tenus par les uns et les autres nous fait davantage encore respecter et aimer les progrès de la médecine, et nous permet aussi de mieux comprendre les raisons pour lesquelles l'innovation tient de Prométhée et de la toile de Pénélope.
À la base des progrès de la médecine, il y a la foi en la science, vérité apparemment toute simple, mais qui mérite d'autant plus d'être rappelée qu'un professeur de médecine a pu déclarer que, jusqu'aux années 1970, le monde médical était resté profondément antiscientifique. Il déplorait ainsi que, dans un grand hôpital parisien, où existait l'un des plus remarquables services d'endocrinologie, la recherche fût absente.
Si, comme l'ont montré les débats d'aujourd'hui, de telles observations ne pourraient plus être émises, c'est précisément parce que les équipes ont faite leur cette déclaration très forte du philosophe anglais Bertrand Russel : « Je crois qu'il faut conserver la conviction que la science est une des gloires de l'homme. Je ne soutiendrai pas que la science n'est jamais dangereuse. Mais ce que je soutiens, et je le dis avec conviction, c'est que la science est plus utile que nuisible et que la peur de connaître est bien plus souvent mauvaise qu'utile ».
À l'évidence, sans cette conviction, aucun saut technologique ne pourrait voir le jour. Car, ainsi que l'a bien résumé le professeur Laurent Degos dans son propos introductif, le saut technologique repose sur les espoirs que suscitent ses bienfaits potentiels, alors même que la société, qui en ignore l'issue, est toutefois prête à en assumer les risques.
Cette configuration triangulaire est celle dans laquelle s'inscrivent les recherches qui nous ont été présentées, qu'il s'agisse, entre autres, de la production en laboratoire de globules rouges à partir de cellules souches dans un but transfusionnel ou de la neuromodulation implantée en psychiatrie.
L'honneur des chercheurs que nous avons entendus ne réside pas seulement dans le souci de promouvoir la santé publique. J'ai en effet relevé qu'ils ne négligent jamais le questionnement éthique, même si certains d'entre eux considèrent que celui-ci, avec l'application du principe de précaution, peut constituer un frein à la recherche, en particulier en ce qui concerne les recherches sur l'embryon.
Il est clair que des controverses sur ce dernier point ne manqueront pas de susciter d'intenses discussions dans les semaines à venir, lorsque l'Assemblée sera prête à débattre de la loi bioéthique.
Quoi qu'il en soit, la nécessité de mettre en place une nouvelle stratégie d'innovation, telle que l'a exposée le Pr. Elias Zerhouni, passe, j'en suis persuadé, par une approche collective, que j'ai appelée la « logique des interfaces » : interfaces entre la communauté scientifique et les décideurs politiques, d'une part, et avec la société civile d'autre part - c'est le rôle que tente de jouer l'Office depuis sa création.
Je ne voudrais pas achever ce discours de clôture sans adresser mes remerciements au secrétariat de l'Office, dont le dévouement et la compétence ont permis à cette remarquable audition publique de se dérouler dans les meilleures conditions.
Je précise qu'un compte rendu intégral de ces tables rondes sera publié et que je présenterai devant l'Office parlementaire une conclusion générale qui sera également publiée.
Les sauts technologiques sont réconfortants pour l'esprit. Mesdames et messieurs, je vous remercie.
EXTRAIT DE LA RÉUNION DE L'OPECST DU 28 JUIN 2011 : PRÉSENTATION DES CONCLUSIONS DE L'AUDITION PUBLIQUE PAR LE PRÉSIDENT CLAUDE BIRRAUX
M. Claude Birraux, président de l'OPECST. L'audition publique sur les sauts technologiques en médecine, qui s'est tenue le 27 janvier 2011, a été suggérée à l'OPECST par le professeur Jean-Michel Dubernard.
Elle s'est proposée d'analyser, à partir de différents exemples, les causes qui sont à l'origine des réussites et des échecs de certains sauts technologiques en médecine.
La France dispose d'excellentes équipes de chercheurs. Toutefois - hormis l'exception remarquable du Professeur Alain Carpentier qui a bénéficié, pour la mise au point du coeur artificiel, d'une relation privilégiée avec Jean-Luc Lagardère - de nombreux projets de recherche ont été ralentis ou n'ont pu aboutir, du fait de certains freins.
Pour surmonter ces derniers, des mesures ont été souhaitées, qui touchent au cadre législatif, aux institutions et au comportement des acteurs.
S'agissant tout d'abord du cadre législatif, il m'apparaît que la concrétisation de certaines des préconisations concernant le droit français et la législation communautaire se heurtent à de sérieuses objections.
Il en est ainsi de la proposition visant à l'abrogation du régime d'interdiction des recherches sur les cellules souches embryonnaires, qui a été regardé comme un frein aux recherches, en matière de thérapie cellulaire et de vitrification ovocytaire.
Les deux Assemblées n'ont toutefois pas souhaité revenir sur le principe de l'interdiction au cours de la discussion du projet de loi relatif à la bioéthique, qui a finalement maintenu le principe d'interdiction avec dérogations, même si la vitrification ovocytaire a été formellement autorisée.
Un second exemple de frein de nature législative aux recherches menées en thérapie génique est imputable non pas au droit français mais au droit communautaire.
Comme le Professeur Jean-Michel Dubernard, il ne m'apparaît pas non plus logique d'assujettir la thérapie génique au régime du médicament. Pour autant, il y a lieu de craindre qu'une réforme ne puisse intervenir dans l'immédiat. Car, ainsi que l'a rappelé M. Jean Marimbert, alors directeur général de l'AFSSAPS, cette situation découle du règlement communautaire du 13 novembre 2007 sur les médicaments de thérapie innovante. Ce texte prévoit une procédure d'autorisation unique dans l'ensemble de l'Union européenne et assimile la thérapie cellulaire à la mise au point d'un médicament. Or, ce faisant, le règlement est allé à l'encontre du régime appliqué en France à la thérapie cellulaire dès 1996-1997, lequel n'était pas exactement calqué sur le régime du médicament.
Une réflexion sur ces dysfonctionnements serait souhaitable, tout comme sur la disparité des pratiques en matière de thérapie cellulaire, relevée par M. Philippe Menasché, directeur de l'unité « Thérapie cellulaire en pathologie cardio-vasculaire » à l'hôpital européen Georges Pompidou. Car, même si l'EMA (Agence européenne du médicament) tente de les uniformiser, les contraintes demeurent différentes selon les États membres, ce qui n'est pas de nature à faciliter les essais multicentriques.
Si l'Europe peut apparaître comme un frein aux sauts technologiques, elle n'en constitue pas moins également un niveau pertinent de réformes. Ainsi serait-il utile, comme l'ont proposé les Professeurs Jean-Michel Dubernard et Laurent Degos, ancien Président de la Haute Autorité de Santé, d'envisager la mise en place d'une instance européenne pour les dispositifs coûteux. On peut, en effet, regretter que, pour ces dispositifs tels que les implants rétiniens, l'Allemagne et la France aient procédé à des essais cliniques sans aucune coopération.
De même, serait-il judicieux de prendre en considération l'idée d'instaurer une procédure européenne d'évaluation formulée par le Professeur Pierre Tiberghien, directeur général délégué à l'EFS. Il s'agirait de veiller à la qualité de l'évaluation par les structures telles que l'AFSSAPS en charge d'autoriser les recherches. Une telle mesure permettrait de réduire les conflits d'intérêts et de garantir une expertise plus pointue.
Au plan institutionnel, j'abonde dans le sens de plusieurs intervenants qui ont déploré le « millefeuille administratif », le maquis de procédures ou encore le parcours du combattant auxquels les chercheurs sont confrontés pour faire aboutir leurs projets. Découragés par une telle situation, certains d'entre eux ont même choisi de s'expatrier.
C'est pourquoi, il m'apparaîtrait nécessaire de se pencher sur deux propositions :
- la première revisiterait la piste déjà explorée du regroupement d'organismes concernés, comme le Comité national consultatif d'éthique, l'Agence de la Biomédecine, l'AFSSAPS, laquelle se trouve justement en cours de restructuration, suite à l'affaire du Médiator ;
- la seconde consisterait à instituer une procédure de guichet unique, distincte des organes de régulation, pour les innovations dans le domaine de la santé ;
La réussite ou l'échec d'un saut technologique tient aussi au comportement des acteurs.
J'ai ainsi pu déplorer dans mon allocution d'ouverture que la médecine de la France souffrait de son caractère administré, ayant fait état des difficultés administratives rencontrées par le professeur Ugo Amaldi, pour développer l'hadronthérapie en France et qui l'ont conduit à retourner dans son pays natal.
A ces difficultés administratives s'ajoute la frilosité des industriels qui, du fait de leur soutien mesuré ou de l'absence de soutien de leur part, ont freiné des projets de recherche ou en ont empêché le développement.
Or, il m'apparaît urgent que les différents acteurs se départissent de tels comportements, afin d'éviter que les atouts réels dont dispose la France ne soient durablement compromis. A cet égard, j'approuve sans réserve les observations de M. Elias Zehrouni, Professeur au Collège de France.
Il a souligné fort opportunément que le besoin d'innovation, qui peut s'analyser comme un facteur négatif en termes de coûts mais positif sur le plan des soins, impose de redéfinir le rapport entre la valeur médicale relative d'une innovation, d'une part, et d'autre part, ses risques, ses bénéfices et ses coûts.
Dans la même perspective, il serait souhaitable que soit entendu l'appel lancé par le Professeur Alain Carpentier aux médecins - souvent taxés, selon lui, d'être dépensiers - par lequel il les exhorte à intégrer dans leur réflexion les conditions économiques et sociales de leurs réalisations.
Il n'était pas sans intérêt d'organiser cette audition publique, d'autant que plusieurs thèmes qui y ont été abordés rejoignent ceux de l'étude menée par Jean-Yves Le Déaut et moi-même sur l'innovation à l'épreuve des peurs et des risques majeurs.
1 Tomographie par émission de positions. Il s'agit d'une méthode d'ingénierie médicale pratiquée par les spécialistes en médecine nucléaire qui permet de mesurer en trois dimensions l'activité métabolique d'un organe.
2 Bonnes pratiques de fabrication : ce sont les pratiques et les systèmes de qualité régissant la fabrication et l'essai de produits pharmaceutiques ou de médicaments.
3 L'hématopoïèse est le processus physiologique permettant la création et le renouvellement des cellules sanguines.
4 Bonnes pratiques de fabrication.